
Abstract
The prevalence and incidence of Parkinson’s disease (PD) is increasing due to a prolonged life expectancy. This highlights the need for a better mechanistic understanding and new therapeutic approaches. However, traditional in vitro and in vivo experimental models to study PD are suboptimal, thus hampering the progress in the field. The epigenetic reprogramming of somatic cells to induced pluripotent stem cells (iPSCs) offers a unique way to overcome this problem, as these cells share many properties of embryonic stem cells (ESCs) including the potential to be transformed into different lineages. PD modeling with iPSCs is nowadays facilitated by the growing availability of high-efficiency neural-specific differentiation protocols and the possibility to correct or induce mutations as well as creating marker cell lines using designer nucleases. These technologies, together with steady advances in human genetics, will likely introduce profound changes in the way we interpret PD and develop new treatments. Here, we summarize the different PD iPSCs reported so far and discuss the challenges for disease modeling using these cell lines.
Keywords
Introduction
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disorder after Alzheimer’s disease. Its incidence increases with age, ultimately affecting ∼1% of the population over the age of 60 and ∼4% over the age of 80.1,2 PD is a chronic progressive disorder characterized by neuronal death in the central nervous system, affecting more significantly the dopaminergic (DA) neurons of the substantia nigra pars compacta (SNpc). Because of the importance of nigrostriatal DA neurons in controlling motor functions, the most noticeable symptoms of PD are bradykynesia, resting tremor, rigidity, and postural instability. Yet, PD is a systemic disorder, and, as the patients’ condition becomes worse, other areas of the brain are affected. 3 Among other consequences, this can cause changes in mood (anxiety, passivity, and depression) and dementia.
The impact of PD on the quality of life is considerable, and the national health costs are alarming, stressing the need for effective therapeutic approaches. So far, there are only two FDA-approved treatments: administration of L-DOPA and deep brain stimulation of the bilateral subthalamic nuclei,4,5 both of which are palliative and not disease modifying. A difficulty for finding curative treatments is that PD only manifests after a big proportion (∼70%) of DA neurons have died. 6 Hence, it is important to identify cohorts at risk and develop preventive measures that stop or delay the disease onset.
Most PD cases are sporadic and idiopathic, resulting from the combination of a permissive genetic background and environmental factors. However, up to 5% of the cases are familial and triggered by known gene mutations. 7 Among these genes, LRRK2, SNCA, PINK1, and PARK2 have been studied in more detail. 8 Mutations in LRRK2 (the most frequent cause of familial PD) and mutations or multiplications of SNCA cause autosomal dominant PD, and in both cases, the underlying mechanism seems to be a gain of function. Yet, these mutations have incomplete (age-dependent) penetrance and are normally associated with late disease onset. Notably, LRRK2 and SNCA are also mutated in a small proportion of sporadic PD patients. On the other hand, loss-of-function mutations in PINK1 and PARK2 cause recessive PD, seem to have full penetrance, and associate with early disease onset.
Understanding the function of PD-related genes is relevant because similar pathways may also participate in idiopathic PD. 9 This has implications at a therapeutic level, as putative drugs effective on a specific group of familial cases may even work on a proportion of idiopathic patients. In this regard, α-synuclein (the product of SNCA) and LRRK2 have been proposed to act on the same molecular pathway, but PINK1 and Parkin (the product of PARK2) seem to work on another. 8 A major pathological mechanism involving α-synuclein is thought to be the deposition of toxic protein aggregates, which in at least some experimental models is LRRK2 dependent. This affects cells by inducing endoplasmic reticulum (ER) stress and/or oxidative stress. Mutant LRRK2 also acts through alternative mechanisms such as changes of protein translation and mitochondrial fragmentation. As for PINK1 and Parkin, the evidence points to a gatekeeper role in regulating mitochondrial homeostasis (clearance, mobility, and fission–fusion dynamics), which, if deregulated, can lead to mitochondrial dysfunction and oxidative stress.
However, despite seminal advances in characterizing the genetic susceptibility to PD, 7 therapeutic developments have been hindered by the lack of optimal in vitro and in vivo experimental models that are predictive of human disease.
PD models
In vitro cell models for PD mostly rely on neuroblastoma cell lines (e.g. SH-SY5Y) that retain the ability to differentiate into DA neurons 10 and PD patient fibroblasts. 11 Yet, the former are transformed cell lines that have the tendency to instability, while fibroblasts have a different gene expression profile and metabolic status compared to neurons. On the other hand, animal models for PD have been set up using worms, flies, rodents, and nonhuman primates. 12 Models for the former three species include either knockouts for genes orthologous to those implicated in hereditary PD or transgene overexpression. Although some of these models show signs of neuronal degeneration, the lack of brain complexity and life span of humans complicate comparisons. As for nonhuman primate PD models, they typically focus on neurotoxins (e.g. MPTP and rotenone) 13 that reproduce the disease manifestations by inducing death in DA neurons but fail to recapitulate the slow chronic progression of PD in humans.
Remarkably, Takahashi and Yamanaka
14
demonstrated in 2006 that retroviral transduction of a cocktail of transcription factors highly enriched in embryonic stem cells (ESCs) could reprogram mouse fibroblasts into ESC-like cells, which they named induced pluripotent stem cells or iPSCs. Multiple groups have subsequently optimized this technology, and it is now possible to produce human iPSCs from diverse donor cell types
15
and using a variety of methods including non-integrating vectors.
16
The differentiation of human iPSCs derived from PD patients into neural cells of interest (DA neurons, other types of neurons, or glia) opens up a new series of exciting possibilities for state-of-the-art in vitro PD modeling that should allow identification of altered signaling pathways and innovative drug screening (Figure 1). The field is in its infancy but a number of reports have already demonstrated proof of principle of such utilities.
17
Schematic showing the potential utility of PD iPSCs for drug discovery and cell transplantation. Human iPSCs from PD patients and healthy people are generated by somatic cell reprogramming. Designer nucleases are used to correct the mutations of PD patient iPSCs or introduce mutations into iPSCs from healthy individuals, thus producing isogenic cell lines. Specific neurons and/or glial cells can be differentiated from those iPSCs and used to study the disease-related phenotypes. Co-culture systems can contribute to studies of non-cell autonomous effects. Once the distinct disease-related phenotypes are characterized, drug-screening platforms can be developed to test compounds that reverse the pathological phenotypes. In the future, putative cell therapy approaches with iPSC-derived neural-like cells will require clinical-grade, good manufacturing protocols or GMP of reprogramming and stringent criteria for iPSC clone selection. (A color version of this figure is available in the online journal.)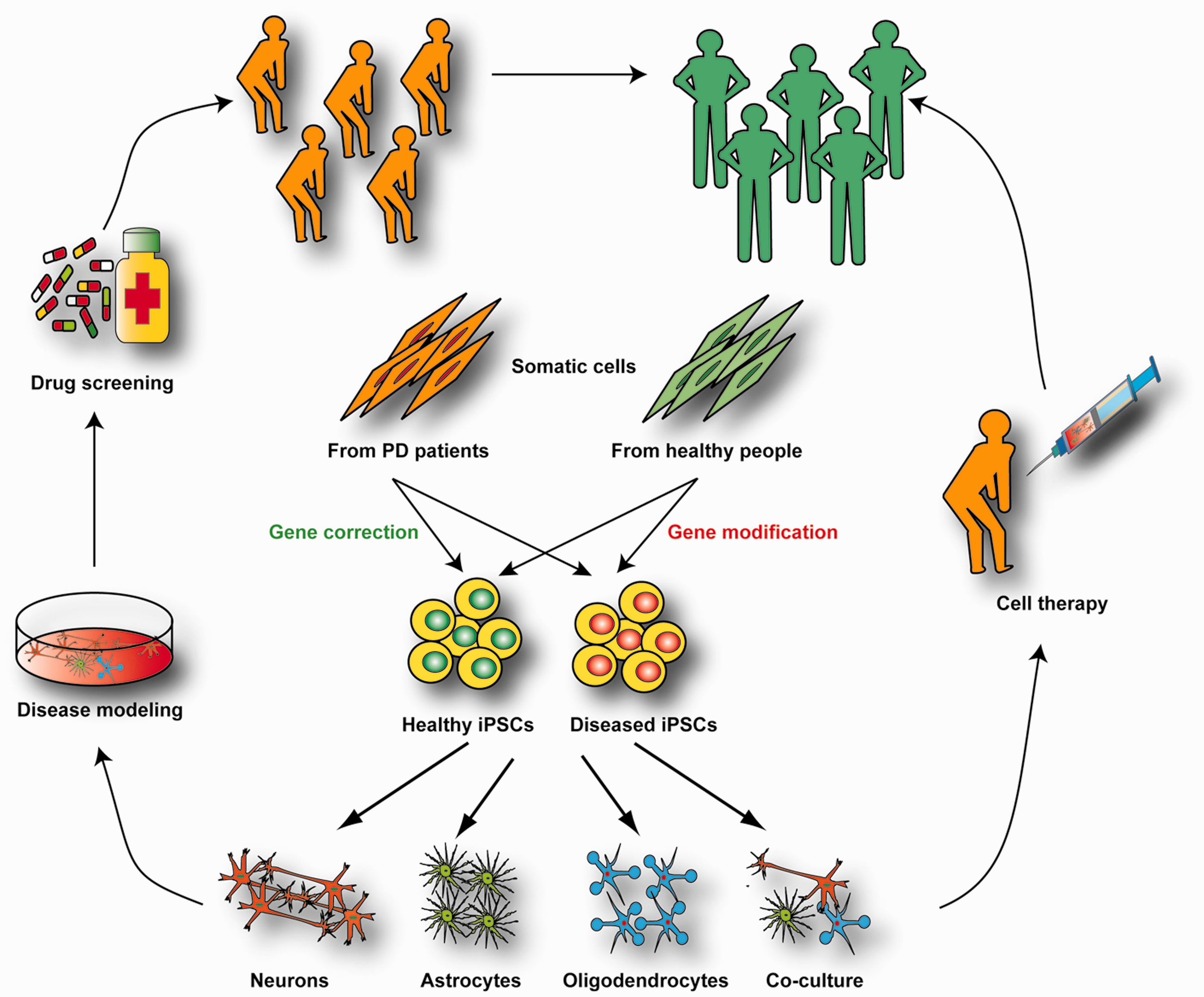
PD in a dish using iPSCs
iPSC models of idiopathic PD
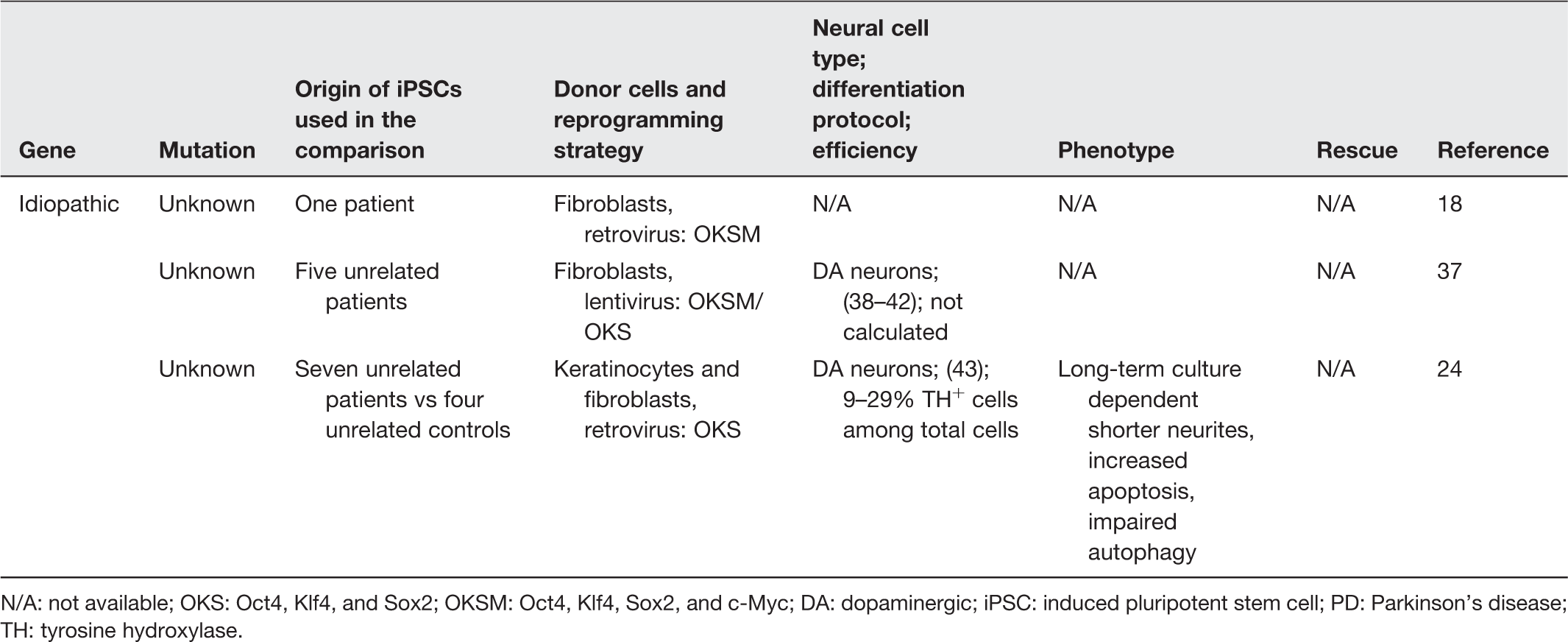
N/A: not available; OKS: Oct4, Klf4, and Sox2; OKSM: Oct4, Klf4, Sox2, and c-Myc; DA: dopaminergic; iPSC: induced pluripotent stem cell; PD: Parkinson’s disease; TH: tyrosine hydroxylase.
iPSC models of autosomal recessive PD

DA: dopaminergic; N/A: not available; NPCs: neural progenitor cells; NURR1: nuclear receptor related 1; OKSMN: Oct4, Klf4, Sox2, c-Myc, and Nanog; OKSMLNshp53: Oct4, Klf4, Sox2, c-Myc, Lin28A, Nanog, and shRNA for p53; iPSC: induced pluripotent stem cell; PD: Parkinson’s disease; TUJ1: neuron-specific class III beta-tubulin.
iPSC models of PD with SNCA mutation
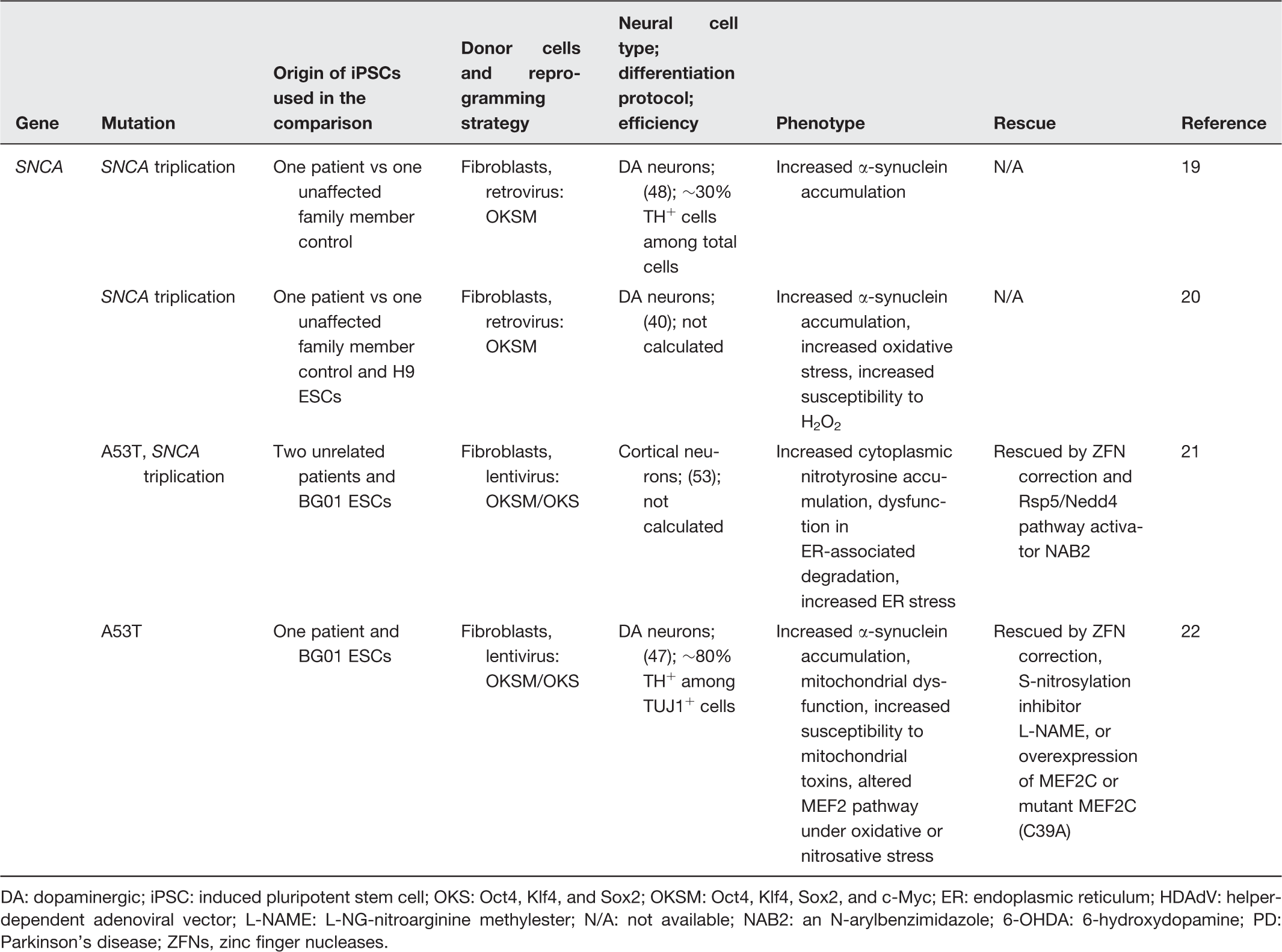
DA: dopaminergic; iPSC: induced pluripotent stem cell; OKS: Oct4, Klf4, and Sox2; OKSM: Oct4, Klf4, Sox2, and c-Myc; ER: endoplasmic reticulum; HDAdV: helper-dependent adenoviral vector; L-NAME: L-NG-nitroarginine methylester; N/A: not available; NAB2: an N-arylbenzimidazole; 6-OHDA: 6-hydroxydopamine; PD: Parkinson’s disease; ZFNs, zinc finger nucleases.
iPSC models of PD with LRRK2 mutation

DA: dopaminergic; iPSC: induced pluripotent stem cell; N/A: not available; NPCs: neural progenitor cells; OKSM: Oct4, Klf4, Sox2, and c-Myc; PD: Parkinson’s disease; TAU: microtubule-associated protein tau; ZFNs: zinc finger nucleases.
Pitfalls of PD modeling with iPSCs
Caveats of PD modeling using iPSCs
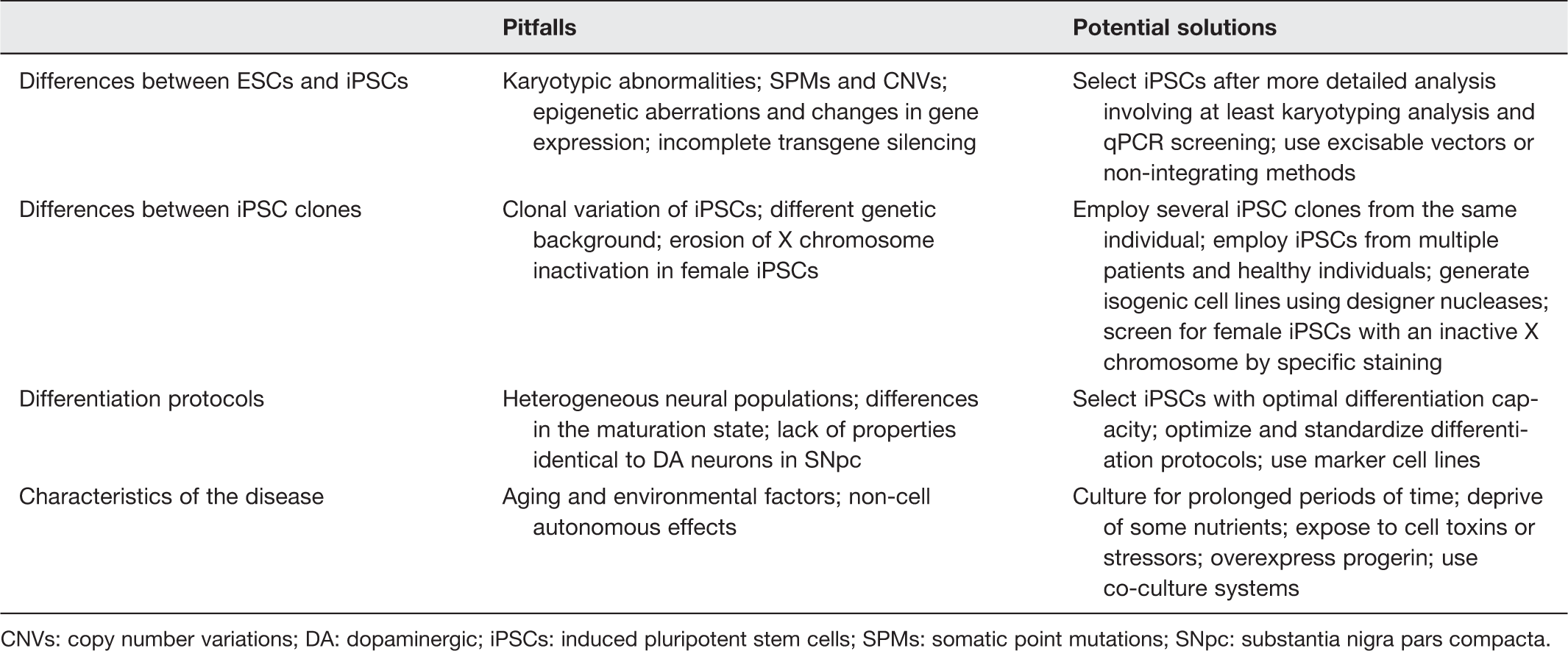
CNVs: copy number variations; DA: dopaminergic; iPSCs: induced pluripotent stem cells; SPMs: somatic point mutations; SNpc: substantia nigra pars compacta.
Differences between ESCs and iPSCs
Reprogramming involves very extensive rearrangement of cellular functions, and consequently, it is prone to errors. 55 Among these errors, there are karyotypic abnormalities, somatic point mutations (SPMs), copy number variations (CNVs), epigenetic aberrations, and variations of gene expression. 56 Importantly, all these alterations can arise during reprogramming but also in the clonal expansion. Karyotyping (e.g. using Giemsa banding) can assess gross abnormalities including aneuploidy and megabase-scale CNVs. Conversely, for detecting SPMs and more subtle CNVs, it is needed to perform genome wide sequencing, which involves higher costs. Nevertheless, compared to karyotypic abnormalities, SPMs and CNVs may not be detrimental for disease modeling unless they affect genes involved in PD. As for the epigenetic aberrations, they can cause variations of gene expression among iPSCs and also influence the propensity to differentiate into given lineages.57,58 However, it must be considered that ESCs show as well a high degree of heterogeneity that is probably determined by the circumstances in which they were derived. 59 Accordingly, the epigenome and gene expression patterns of some iPSCs are closer to the average ESC than other ESCs may be, 60 and with adequate screening, it is possible to select iPSCs that perform equally well in differentiation assays compared to optimal ESCs.60,61
Differences between iPSC clones
Differences between ESCs and iPSCs are likely not an overwhelming drawback for PD modeling, but differences between individual iPSC clones might be if not handled properly. This is supported by the observation that iPSC clones produced from the same individual (even in the same reprogramming experiment) can have dissimilar characteristics including those that influence neural differentiation performance. 62 Notably, this problem becomes even more relevant when iPSCs from a given PD patient are compared with those from other patients or healthy controls; as in this scenario, the genetic background introduces a new source of variability. Contrasting large numbers of iPSCs from PD patients with those from healthy age-matched individuals can potentially minimize this caveat. Yet, such endeavor may not only be impractical due to high costs and manpower limitations, but also potentially misleading if we consider that healthy controls may also develop PD (or another neurodegenerative disease) at a later time point. This consideration is particularly important for those families affected by PD in which some individuals have developed the symptoms while others have not. A possible solution for allowing authentic comparisons among diseased and healthy iPSCs is to correct the corresponding PD gene mutations with designer nucleases: zinc fingers nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats associated nuclease Cas9 or CRISPR-Cas9. 63 In this regard, Soldner et al. 64 reported the first isogenic iPSC model generated with this methodology, which involved correcting the A53T substitution in α-synuclein in PD iPSCs using ZFNs. Ever since, the number of reports using isogenic iPSCs has increased steadily, and this will likely become a standard approach in the field.21,22,26,27,30 Nevertheless, gene editing with designer nucleases has the risk of off-target effects65,66 and requires lengthy cell expansion that may increase the number of passages to a point in which the iPSCs become unstable. 59 Hence, exhaustive screening of iPSCs modified with designer nucleases is also necessary before comparative analysis. Moreover, it can only be applied to those cases (familial or sporadic) where the mutations are known.
Two other potential caveats intrinsic to PD modeling with iPSCs are the gender and the use of integrating vectors. Regarding the former caveat, it is known that female iPSCs can undergo erosion of X chromosome inactivation leading to altered expression of genes linked to cognition and brain development. 67 However, selection of optimal iPSCs with a relatively simple screening procedure such as immunofluorescence for the repressive histone mark H3K27me3 can help overcome this concern. 68 As for the latter caveat, suboptimal ESC-like properties and reduced differentiation potential of iPSCs may also be caused by incomplete transgene silencing if the iPSCs were obtained by means of viral transduction. 37 This can be solved with proper PCR screening and using excisable vectors or non-integrating delivery methods (e.g. episomal). 69
Differentiation protocols
A major problem of existing protocols for differentiation into any neuronal cell types (including DA neurons) is that they yield heterogeneous populations consisting of both neurons maturating at different time points and non-neuronal cell types (progenitor cells, glia, and other intermediates). 62 This can introduce significant variability when detecting in vitro disease phenotypes for three different reasons. First, in PD patients, DA neurons (in particular those from the SNpc) are more vulnerable to cell death than other cell types. 1 Therefore, a phenotype that appears in DA neurons may not occur in a predominantly non-DA neuronal population and vice versa. Second, DA neurons (and other neuronal cell types as well) maturating at different speeds could be confounded with morphological abnormalities (e.g. in dendrites) belonging to a disease phenotype. The latter could be caused for example by variations (even if small) in the propensity to differentiation of independent iPSC clones. Selecting iPSC clones with comparable differentiation capacity may eliminate this problem but could also select against potential developmental defects induced by certain gene mutations. 70 In this regard, Liu et al. 26 described eliminate defects in iPSC-derived NPCs from PD patients with mutant LRRK2. 26 Third, other neural cell types besides neurons (e.g. astrocytes or microglia) are affected in PD and may contribute as well to the in vitro phenotype.71,72 Of note, high-efficiency protocols for DA neuronal differentiation 47 may eliminate the interference produced by other cell types (neuronal or non-neuronal), but at the same time block a non-cell autonomous effect (e.g. toxic substances from glia affecting neurons) necessary for the in vitro phenotype. 73 Creating marker cell lines by inserting a reporter cassette (e.g. GFP and antibiotic resistance genes) into a given locus (e.g. dopamine transporter or DAT locus) with designer nucleases is a potential solution, 74 as this can allow the detection of disease phenotypes specifically on those cells of interest while non-cell autonomous effects are maintained. In the future, co-culture of different iPSC-derived neural cell types will be important as well to help discern cell autonomous and non-cell autonomous effects. Another relevant issue is that existing protocols for DA neuronal differentiation are time consuming. To solve this issue, directed differentiation of iPSCs with specific transcription factors is emerging as a promising alternative.75,76
Characteristics of the disease
PD is a chronic disease in which environmental factors and age play key roles. 77 The latter is especially relevant in idiopathic cases but also applies to PD patients bearing mutations in LRRK2 or SNCA. Hence, it is relevant to reproduce these circumstances in vitro in order to model PD with iPSCs-derived neural cells more faithfully. Two simple methods to mimic stress and aging are culturing iPSCs-derived neural cells for prolonged periods of time 24 and depriving them of essential nutrients. 27 In addition, other groups have employed chemicals such as pro-oxidants,22,23,25,27 ER stressors, 21 mitochondrial depolarizing drugs,25,31,35 and proteasomal inhibitors23,25 not present in standard conditions to induce disease phenotypes. Likewise, Miller et al. 33 overexpressed a mutant form of lamin A (progerin) responsible for accelerated aging in Hutchinson Gilford Progeria patients to induce an age-related phenotype in PD iPSC-derived neurons.
Conclusions and future perspectives
With the arrival of the iPSC technology, we can do patient-specific PD modeling using neural cells that are more similar to those affected in PD patients in vivo. However, this method is not exempt of concerns and requires careful considerations.
70
Besides the caveats explained above, PD modeling in a dish lacks many aspects of brain complexity and thus could be misleading. Interestingly, Lancaster et al.
78
generated human brain-like structures termed cerebral organoids by embedding human iPSCs in a three-dimensional organoid culture system, which raises hope for capturing some of the complexity of human brain in vitro. Likewise, a proper understanding of PD with iPSCs will require the generation of large numbers of iPSC clones from patients with known gene mutations and idiopathic cases, which is a problematic task for any single laboratory. This endeavor may thus be achieved more easily as part of research consortia, in which case, it will be important to standardize all procedures (for reprogramming and also the subsequent expansion). An attractive alternative is to complement research on PD using neural cells produced by means of somatic cells transdifferentiation.75,76,79 The latter has the advantage that the procedure is quicker, and the costs are smaller, thus allowing simultaneous manipulation of many samples. However, so far, neural transdifferentiation protocols are in general inefficient. Besides, large numbers of primary cells are needed and genome engineering is less amenable. In summary, iPSC-based PD models have a promising future if the exiting caveats are overcome, and this will hopefully open new avenues for mechanistic studies, drug discovery, and clinical therapy of PD (Figure 2).
PD iPSCs are proving useful to fill in some of the missing pieces of the PD puzzle. So far, PD iPSC models have been utilized to gain insights into the role of autophagy, mitochondrial homeostasis, MEF2 pathway, ERK pathway, and ER stress in PD. (A color version of this figure is available in the online journal.)
Footnotes
Author contributions
PZ and ZL contributed equally to this work. PZ, ZL, WF and MAE wrote the manuscript; JY, DPI, ZH and MDT helped edit the manuscript; PZ, ZL, WF and WT prepared the main figures and tables; MAE coordinated the entire process and approved the final version.
ACKNOWLEDEGMENTS
Work on this topic in the Esteban laboratory is supported by grants from the Strategic Priority Research Program of the Chinese Academy of Sciences (number XDA01020106), the Ministry of Science and Technology of China 973 program (2011CB965200) and the Queensland-Chinese Academy of Sciences (Q-CAS) Biotechnology Fund (GJHZ1242).