
Abstract
ABT-737 is a BH3-mimetic that has a wide spectrum of single-agent activity against acute lymphoblastic leukemia (ALL) cell lines and xenografts. Previously, we reported that in response to ABT-737, ABT-737-resistant ALL cell lines showed an apparent increase in Mcl-1 (an anti-apoptotic Bcl-2 family protein that is not effectively inhibited by ABT-737) while ABT-737-sensitive ALL cell lines showed decreased Mcl-1 levels. Here we explored the mechanism of Mcl-1 cleavage by ABT-737 and the effect of adjacent phosphorylation sites on Mcl-1 cleavage and apoptosis induced by ABT-737 in a human B-lineage ALL cell line. Caspase cleavage sites in Mcl-1 and the effect of mutation in Mcl-1 phosphorylation sites were determined by transducing Mcl-1 variants tagged with the V5 epitope into human ALL cells. Cytotoxicity was by fluorescence-based DIMSCAN, and changes in protein by immunoblotting. ABT-737 induced a caspase-dependent cleavage of Mcl-1. Of the two Mcl-1 caspase cleavage sites (D127 and D157), D157 was the site of ABT-737-induced cleavage in ALL cells. Cells with exogenously expressed Mcl-1 Δ157 fragment showed greater caspase-3 and caspase-9 activation when they were treated with ABT-737 compared with cells expressing wild-type or D157A mutant Mcl-1. Cells with mutated phosphorylation sites on Mcl-1 (S159A and T163A) were less susceptible to Mcl-1 cleavage and apoptosis induced by ABT-737. Our data showed that Mcl-1 is post-translationally regulated in response to ABT-737 treatment, primarily via a caspase-dependent cleavage that generates a pro-apoptotic Mcl-1 fragment.
Introduction
The Bcl-2 family of proteins consist of 25 pro- and anti-apoptotic members that interact to maintain a balance between newly forming cells and old dying cells. 1 The interaction of the Bcl-2 family of proteins determines apoptotic cell death in response to cancer therapy. Targeting the anti-apoptotic Bcl-2 family of proteins can potentially alter apoptotic “thresholds” and overcome drug resistance to cancer chemotherapy. Constitutively high levels of Bcl-2 or Bcl-XL have been associated with a more aggressive malignant phenotype and/or drug resistance to various categories of chemotherapeutic agents in both hematologic malignancies and solid tumors.2–9 Overexpression of Bcl-2 RNA and/or protein has been observed in acute myelogenous leukemia (AML) and in acute lymphoblastic leukemia (ALL),10,11 and the Bax/Bcl-2 ratio inversely correlates with prognosis of AML and ALL.12,13 Bcl-2 overexpression is commonly observed, but is highly variable at diagnosis in ALL.3,10,11,14 In ALL, both Bcl-XL overexpression 2 and the ratio of Bcl-2 to the proapoptotic Bcl-2 family protein Bax 15 have been inversely associated with prognosis and high Bcl-2 expression has been associated with decreased responsiveness to induction chemotherapy. 16 Thus, the anti-apoptotic Bcl-2 family of proteins provide an attractive therapeutic target for ALL.
Initial therapeutic approaches to targeting Bcl-2 employed a Bcl-2 antisense (Oblimersen sodium), which has been tested in phase III studies, and a number of small-molecule inhibitors of the anti-apoptotic Bcl-2 family proteins are currently in preclinical or clinical stages of development. 1 ABT-737 is one of the small-molecule inhibitors that is designed to mimic the direct binding of a pro-apoptotic Bcl-2 protein (e.g. Bad) to the hydrophobic groove of anti-apoptotic Bcl-2 family proteins, including Bcl-2, Bcl-XL, and Bcl-w. 17 ABT-737 inhibits pro-apoptotic Bcl-2 proteins such as Bad, Bid, and Bim (direct activators of Bax or Bak), from forming heterodimers with anti-apoptotic Bcl-2 family proteins, thereby promoting Bax and Bak oligomerization. 17 ABT-737 markedly increases the cytotoxic response of cancer cells to radiation and has shown preclinical activity as a single agent or in combination with other chemotherapeutic agents against AML,18,19 multiple myeloma, 20 lymphoma, 21 chronic lymphocytic leukemia, 22 small-cell lung cancer,17,23,24 ALL, 25 and neuroblastoma.26,27 Some Bcl-2 family members, namely the myeloid cell leukemia 1 (Mcl-1) protein and branched floretless 1 (Bfl-1, also called A1, BFL-1/A1, Bcl2A1) have relatively low binding affinity for ABT-737. 17 High constitutive levels of Mcl-1 expression in small-cell lung cancer cells 23 and in other types of cancer cells have been associated with resistance to ABT-737.22,28–30
Responses to ABT-263 (navitoclax, an orally available Bad mimetic related to ABT-737) as a single agent in early phase clinical trials were less than expected31,32 based on the promising preclinical data.17,21,23,24 While the ultimate clinical application for BH3 mimetic drugs may lie in appropriate drug combinations,25,33 it is important to define the molecular mechanisms that determine the single agent sensitivity to the ABT-737 and similar agents. As mentioned previously, multiple reports have suggested that high constitutive levels of Mcl-1 expression are associated with resistance to ABT-737.19,21,22,29,30 In acute lymphoid malignancies, we demonstrated that a reactionary increase in Mcl-1 protein expression induced by ABT-737, rather than constitutive levels of Mcl-1 expression, was associated with resistance to ABT-737. We also observed ABT-737 sensitivity was invariably accompanied by an apparent decrease in Mcl-1 protein levels. 33 In the current study, we investigated the mechanisms of Mcl-1 cleavage in response to ABT-737 treatment.
Materials and methods
Chemicals
ABT-737 was supplied by Abbott Laboratories, Abbott Park, IL. Dimethylacetamide (DMA), 1,4-dithio-
Cell culture
Human ALL cell lines COG-LL-329, COG-LL-332, COG-LL-317 (human T-cell ALL), and COG-LL-319 (human pre-B leukemia) cell lines were established by the Children's Oncology Group (COG) Cell Line and Xenograft Repository (www.COGcell.org) as previously described
34
from clinical samples that were provided by the COG under written informed consent. COG cell lines were maintained in Iscove's modified Dulbecco's medium (Cambrex, Walkersville, MD) supplemented with 3 mmol/L
Mycoplasma testing was performed at the Molecular Biology Core Lab, Cancer Center, School of Medicine, Texas Tech University Health Sciences Center (Lubbock, TX) and all cell lines used were mycoplasma-free. Cell line identities were confirmed by short tandem repeat profiling as previously described 35 ; short tandem repeats were unique for all cell lines and were verified against original profiles in the COG STR database (www.COGcell.org).
Generation of mutant constructs and stable transfection/transduction
Stable transfection
To investigate the cleavage site of Mcl-1, V5-tagged Mcl-1, Mcl-1D127A, and Mcl-1D157A were expressed in CCRF-CEM, NALM-6, HEK-293FT, or RS4;11 cells. Point mutations were introduced to two caspase-cleavage sites (D127 and D157) of Mcl-1 using QuickChange Lightning Site-Directed Mutagenesis Kit (Stratagene Cloning Systems, La Jolla, CA) with primers (Supplementary Figure 1b, primers 1–4) and confirmed by sequencing. dsDNA templates with or without mutations were cloned into the p3XFLAG-myc-CMV26 vector (Sigma) after digestion with HindIII and EcoRI (New England Biolabs). The recombinant p3XFLAG-myc-CMV26-Mcl-1 plasmids (Supplementary Figure 1c) were transfected into CCRF-CEM cells using electroporation as described previously. 33 Stable transformants were selected using G418 (Life Technologies).
Lentiviral vector transduction
Lentiviral vectors were used to identify Mcl-1 cleavage site(s) in response to ABT-737 treatment and to determine the role of phosphorylation on Mcl-1 cleavage. Each V5-tagged Mcl-1 (wild-type; mt on caspase-cleavage sites [D127A and D157A]; mt on phosphorylation sites [S159A and T163A]) was amplified using primers (Supplementary Figure 1b, primers 9–12) and confirmed by sequencing. The PCR products with or without mutation were cloned in pDONR™221 vector (Life Technologies). Recombination of pEntry-Mcl-1 and pLenti6.3/V5-DEST was performed to generate the pLenti6.3/V5 expression construct (Figure 3a). HEK-293FT cells were cotransfected with pLenti6.3/V5 expression construct and the optimized packaging mix (pLP1, pLP2, and pLP/VSVG plasmids, at 1 μg/μL in TE buffer, pH 8.0, Invitrogen Life Technologies, Carlsbad, CA). Viral supernatant was harvested at 48 h after transfection, and the titer was determined as described by the manufacturer. The viral supernatant (1.0 × 107 infective particles/mL) was added to NALM-6 cells. Stably transduced cells were selected using blasticidin (5 μg/mL). Cells were then analyzed for V5 by Western blotting using an antibody against V5 (Abcam). To investigate the effect of the mutation on the caspase cleavage sites in Mcl-1 following ABT-737 treatment, we seeded cells stably transduced with or without mutation in six-well plates, treated the cells with ABT-737 (10 μmol/L for 24 h), and assessed the cell death by DIMSCAN analysis.
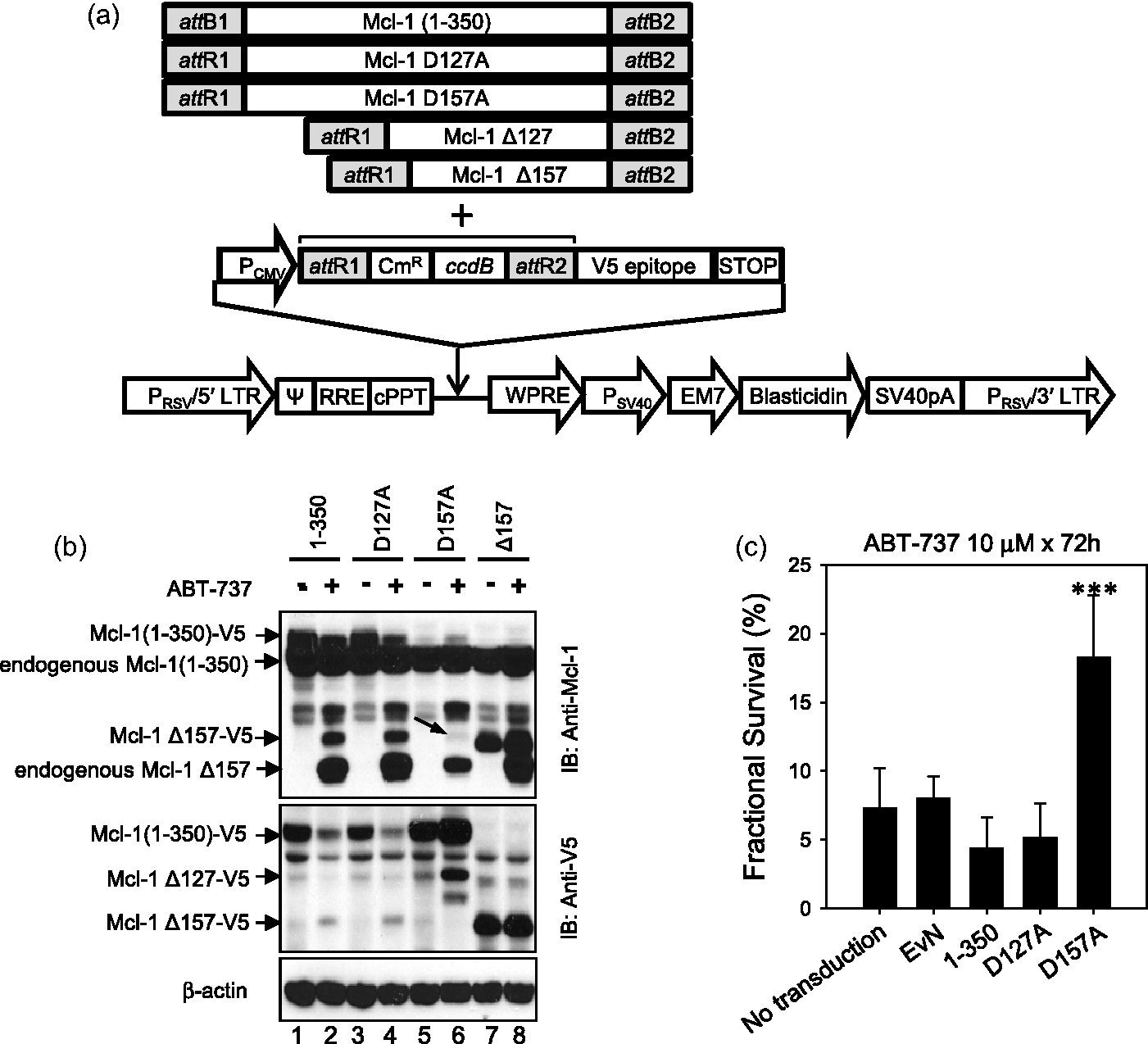
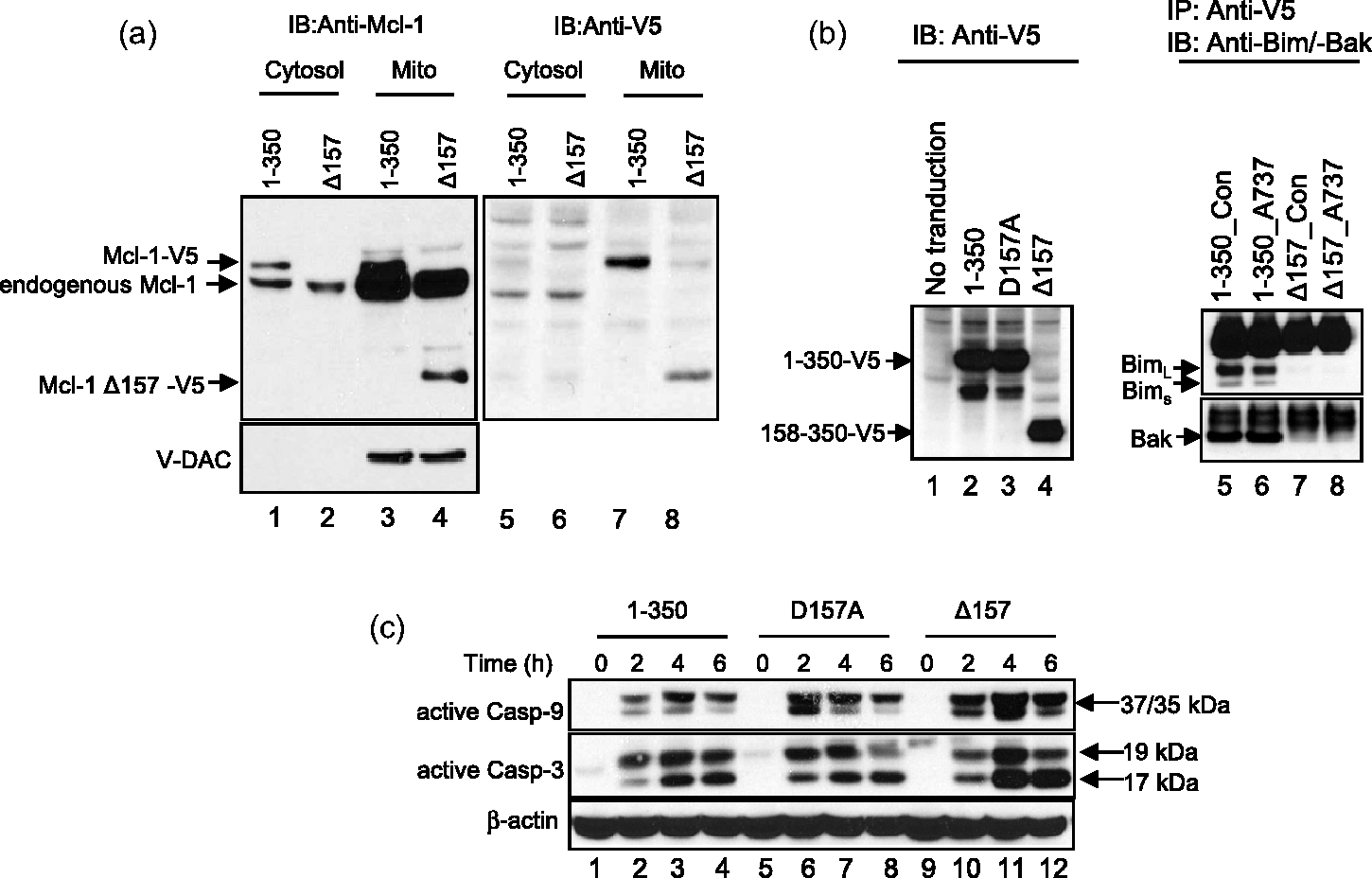
Caspase-dependent Mcl-1 cleavage by ABT-737 in ALL cell lines. (a) Mcl-1 cleavage after ABT-737 treatment. Changes in Mcl-1 protein was assessed after the treatment with ABT-737 in two relatively resistant (NALM-6 and CCRF-CEM) and two relatively sensitive (COG-LL-319 and RS4;11) cell lines. At achievable levels of ABT-737 in vivo (up to 1 μmol/L in NALM-6 and CCRF-CEM and 100 nmol/L in COG-LL-319 and RS4;11 for 6 h), the resistant cell lines did not show cleavage of Mcl-1 after incubation with the drug. After treatment, cells were used to prepare whole cell lysates, which were subjected to immunoblot analysis with a monoclonal antibody to Mcl-1. Immunoblotting for β-actin was used as a control for equal protein loading. Data shown are representative of those obtained in three separate experiments. A horizontal line has been inserted in RS4;11 data to indicate different antibodies used for uncleaved Mcl-1 (cat # 4572, Cell Signaling Technology) and cleaved Mcl-1 (cat # 5453, Cell Signaling Technology). The data from another experiment of the same samples were used to better illustrate the changes in uncleaved form of Mcl-1 in RS4;11 cells. The rest of the blotting in the figure used Mcl-1 antibody (cat # 5453). (b and c) Caspase-dependent cleavage of Mcl-1 in ALL cell lines. To investigate by which mechanisms Mcl-1 is cleaved, one resistant (NALM-6, b) and one sensitive (COG-LL-319, c) cell lines were utilized for the experiment. NALM-6 treated with higher concentrations (10 μmol/L) than in vivo achievable concentrations showed cleavage of Mcl-1 by ABT-737 treatment. Both NALM-6 and COG-LL-319 cells were pre-incubated with either 80 μmol/L Boc-d-fmk, a pan caspase inhibitor, or 10 μmol/L MG-132 for 2 h before cells were treated with ABT-737 (10 μmol/L for NALM-6 and 200 nmol/L for COG-LL-319) for 6 h. Changes in Mcl-1 were assessed by immunoblotting. β-Actin was used as a control for equal protein loading. Data are representative of three experiments Mcl-1 D157 as a caspase cleavage site after ABT-737 treatment. (a) pLenti expression constructs containing Mcl-1 variants used for lentiviral transduction. Mcl-1 plasmids were constructed using pLenti6.3/V5-DEST. All Mcl-1 PCR products were amplified using specific PCR primers listed in Supplementary Figure 1b (#5–8) and each product contains attR1 and attR2 recombination sites. (b) Mcl-1 D157 as a caspase cleavage site by ABT-737. Caspase cleavage sites in Mcl-1 after ABT-737 treatment were investigated in NALM-6 cells transduced with lentiviral vectors. To identify the cleavage site in Mcl-1, cells exogenously expressing Mcl-1-V5 variants (1-350, D127A, D157A, 158-350: Δ157), were treated with ABT-737 10 μmol/L. Mcl-1 cleavage was detected by immunoblotting against Mcl-1 and V5 as described in “Materials and methods” section. (c) Changes in cytotoxicity of ABT-737 in cells with mutated cleavage sites. NALM-6 cells untransduced or stably expressing empty vector (EvN), Mcl-1 full length (1–350), Mcl-1 with aspartate127 mutation (D127A), or Mcl-1 with aspartate157 mutation (D157A) were incubated with 10 μmol/L ABT-737 for 72 h, and the cytotoxicities were assessed using the DIMSCAN system (n = 6, ***P < 0.001) Localization and interaction of Mcl-1 fragment. (a) Localization of Mcl-1 fragment (Δ157). Cytosol and mitochondria were fractionated in cells expressing full-length Mcl-1 tagged with V5 and Mcl-1 fragment (Δ157). Then, the fractionated samples were subjected to immunoblot analysis with a monoclonal antibody to Mcl-1 (left) and V5 (right). V-DAC was used to confirm the isolation of cytosol fraction without mitochondrial contamination. (b) Interaction of Mcl-1 Δ157 fragment with other pro-apoptotic Bcl-2 family of proteins. Interaction between C-terminal fragment of Mcl-1 and proapoptotic Bcl-2 family of proteins (Bim and Bak). To investigate whether the N-terminal domain of Mcl-1 is required to bind with pro-apoptotic Bcl-2 proteins (Bim and Bak), HEK-293FT cells were transiently transfected with V5-tagged Mcl-1 (1–350 or 158–350: Δ157). Immunoprecipitations with 5 µg anti-V5 antibody were performed and the presence of immunocomplexes was determined by immunoblotting. (c) Mcl-1 effect on caspase activation. Full length (1–350), D157A, and Δ157 were incubated with 10 μmol/L ABT-737 and caspase activation was analyzed every 2 h by immunoblotting. β-Actin was used as a control for equal protein loading. The data presented were reproducible in a repeat experiment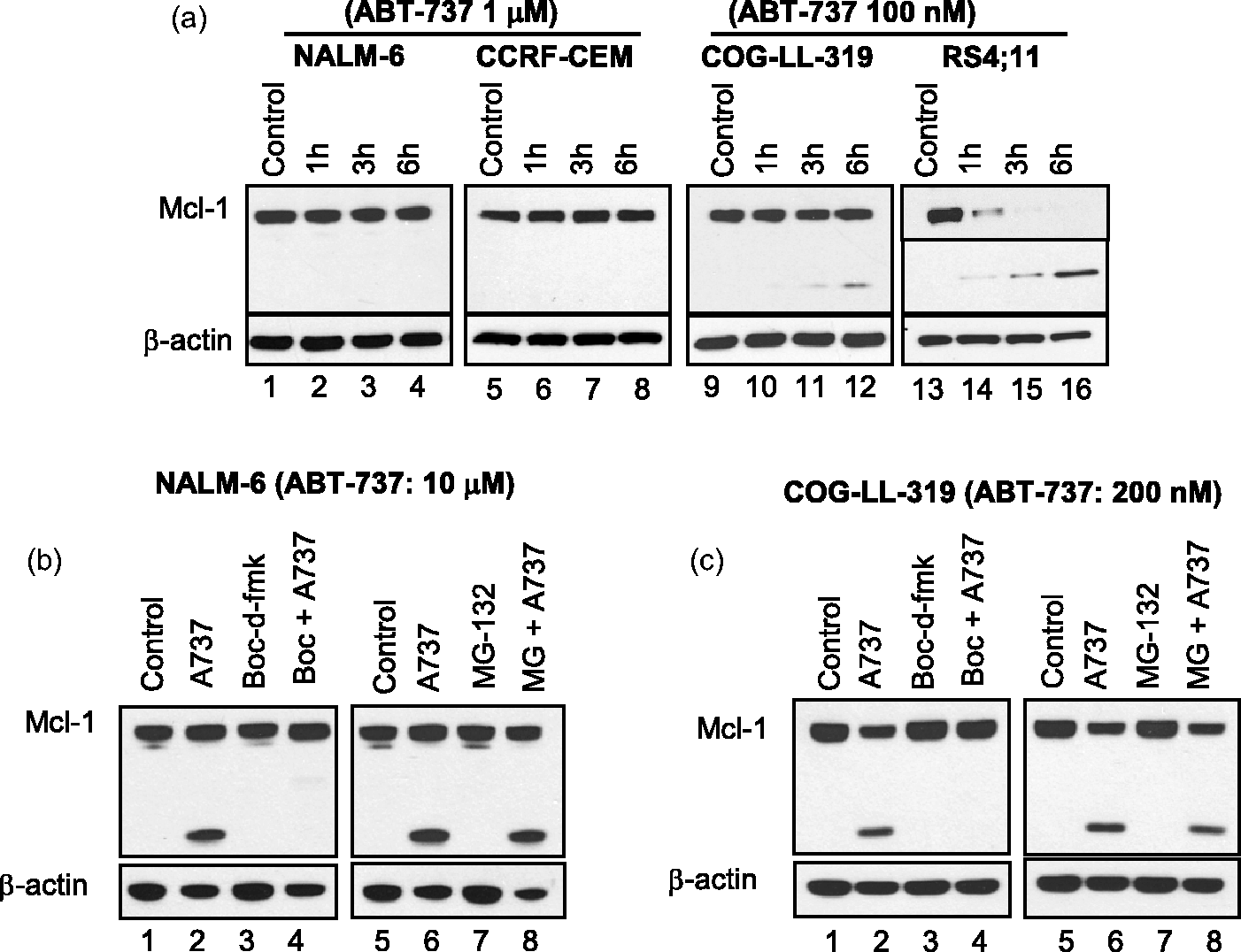
Real-time reverse transcription-polymerase chain reaction
We used real-time reverse transcription-polymerase chain reaction (RT-PCR) and an ABI 7900 HT Fast RT-PCR System (Applied Biosystems, Foster City, CA) to quantify basal mRNA expression of STK31 in each of the seven ALL cell lines used. Primers and probe (STK31-F: 5′-AGC TGC CTC TGC TTC ATC CTG AAA-3′, STK31-R: 5′-AGT TCC TTC ATG GGC TCA GCA TCA-3′, STK31-probe: 5′-TGG TGG TCT CCT TAC AAT GAG CTT GG-3′) were designed and synthesized with the use of PrimerQuest software (Integrated DNA Technologies) using sequences obtained from the GenBank database of Integrated DNA Technologies. The probes were labeled on the 5′ nucleotide with the fluorescent reporter dye 6-carboxy-fluorescein and on the 3′ nucleotide with the fluorescent quenching dye 6-carboxy-tetramethyl-rhodamine. The primers were designed so that the amplicon spanned at least two exons to avoid amplification of genomic DNA.
All RT-PCR assays were performed in triplicate in 96-well plates using Master Mix Reagents (TaqMan one-step RT-PCR Master Mix Reagents kit; Applied Biosystems), 200 nmol/L forward primer, 200 nmol/L reverse primer, 100 nmol/L of probe, and 100 ng of total RNA in a total volume of 25 μL. Total RNA was extracted from each of the seven ALL cell lines with the use of TRIzol reagent (Invitrogen Life Technologies). The cycling conditions were 30 min at 48℃ for reverse transcription, 10 min at 95℃ for initial activation, and 40 cycles of 15 s at 95℃ and 60℃ for 1 min. TaqMan real-time RT-PCR data were analyzed with the use of Sequence Detector V1.7 software (Perkin-Elmer Applied Biosystems, Foster City, CA). The RNA level for each sample was normalized to the 18S rRNA level, which was measured on the same plate.
Cytotoxicity assay
Cytotoxic effects of ABT-737 in ALL cell lines with or without mutated Mcl-1 were assessed using DIMSCAN, a semiautomatic fluorescence-based digital image microscopy system that quantifies viable cells in tissue culture multiwell plates on the basis of their selective accumulation of fluorescein diacetate (FDA).36,37 Cells were seeded into 96-well plates in 150 μL of complete medium (15,000 cells per well) and incubated for 16–24 h. ABT-737 (stock solution: 10 mmol/L ABT-737 in DMSO) was added in 50 μL of culture medium per well at appropriate concentrations, and the cells were incubated at 37℃ in 12 replicates for 48–72 h. Each assay was repeated twice. We then added 50 μL of FDA in 0.5% eosin Y to each well (final concentrations: FDA = 10 μg/mL, eosin Y = 0.1% [w/v]) and incubated the cells for 15 min at 37℃. Total fluorescence in each well was measured with the use of DIMSCAN, and the results were expressed as surviving fractions of treated cells compared with control cells that were exposed to vehicle (either 0.1% DMSO or ethanol in medium).
Immunoblot assay and immunoprecipitation
Cells were lysed in radioimmunoprecipitation (RIPA) lysis buffer consisting of 10% (vol/vol) 10x RIPA lysis buffer (Millipore), 1 mmol/L phenylmethanesulphonylfluoride (PMSF), 1 mmol/L sodium orthovanadate, 1 mmol/L sodium fluoride, 1 µg/mL aprotinin, 1 µg/mL leupeptin, and 1 µg/mL pepstatin. The lysates were incubated on ice for 15 min, sonicated briefly, and centrifuged at 12,000 × g for 15 min. The amount of protein in the supernatants was determined using the bicinchoninic acid (BCA) protein assay kit (Pierce Biotechnology, Rockford, IL), and equal amounts of protein were resolved by electrophoresis on a 4–12% NuPAGE® Bis-Tris gel (Invitrogen Life Technologies) and transferred to a nitrocellulose membrane (Protran, Keene, NH), and the membrane was incubated with primary antibodies (rabbit monoclonal anti-Mcl-1 (Cell Signaling Technology), anti-capase-3, -8, -9 (Cell Signaling Technology), anti-V5 (Abcam), or anti-Bim (Cell signaling)) at 1:1000 dilution, followed by horseradish peroxidase (HRP)-conjugated secondary antibodies (anti-mouse IgG (Cell Signaling Technology) or anti-rabbit (Cell Signaling Technology) at 1:2000 dilution. Antibody binding was then detected with the use of a chemiluminescent substrate (GE Healthcare) and visualized on autoradiography film (Denville Scientific, Inc).
To examine protein–protein interactions between pro-apoptotic Bcl-2 members and various lengths of Mcl-1 (1–350, 128–350 or 158–350 amino acids), we used the Dynabeads® Immunoprecipitation kit (Invitrogen Life Technologies). Wild-type Mcl-1 and Mcl-1 fragments tagged with V5 were expressed in HEK-293FT cells, and the lysates were collected. HEK-293FT cells were used for the experiments as Bax expression in NALM-6 cells is minimal. Anti-V5 antibody was added in 1.5 mL microtubes containing Dynabeads Protein G. Then, the lysate was added to react with antibody, followed by denaturing elution for isolation of denatured target protein, and immunoblotting as described above.
Apoptosis assay
Assessment of apoptosis by flow cytometry
Apoptosis was evaluated by assessing the subdiploid DNA content and Annexin-V binding, which detects phosphatidylserine inversion in cells that are undergoing apoptosis, using flow cytometry. NALM-6 and RS4;11 cells were treated with vehicle control or ABT-737, washed twice with ice-cold phosphate-buffered saline (PBS), and resuspended in 100 μL of binding buffer (10 mmol/L HEPES-NaOH [pH7.4], 140 mmol/L NaCl, 25 mmol/L CaCl2) at a final concentration of 0.5 × 106 cells/50 μL. Annexin-V conjugated with fluorescein (Annexin-V-FITC: 5 μL; BD Biosciences) was added to the cell suspension and the mixture was incubated for 10 min at room temperature. The cells were washed and resuspended in 500 μL of binding buffer. Just prior to flow cytometry analysis, 3 μL of a 50-μg/mL propidium iodide (PI) stock solution was added for counterstaining to distinguish cells in the early stage of apoptosis (Annexin-V-positive cells) from cells that have a subdiploid DNA content (PI-positive) and that are therefore considered to be in the late stage of apoptosis. A BD LSR II flow cytometer was used (BD Biosciences), operated with DiVa (version 4.1.2) software. The cells were analyzed by flow cytometry with band pass filters of 525 ± 25 nm for FITC and 610 ± 20 nm for PI.
Caspase-3 activation
Capase-3 activity assay kit (Cell Signaling) was used to assess the enzyme activation according to the manufacturer's instructions. The cells were incubated with vehicle or ABT-737 for 0 or 4 h, and the cells were lysed to collect the supernatant. Protein concentration in the supernatant was quantitated using a BCA protein assay kit. 10 μL of supernatants were transferred to a black 96-well plate, and 200 μL of solution that contains fluorogenic substrate (N-acetyl-Asp-Glu-Val-Asp-7-amino-4-methylcoumarin or Ac-DEVD-AMC) for caspase-3. After 1 and 4 h of incubation, relative fluorescence units (RFUs) were measured on a fluorescence plate reader (Spectramax M2e, Molecular Devices) with excitation at 380 nm and emission at 480 nm. The RFU values were normalized by the amount of protein.
Oligonucleotide array expression analysis
Total RNA was isolated from COG-LL-317, COG-LL-319, COG-LL-329, COG-LL-332, NALM-6, MOLT-3, MOLT-4, and RS4;11 cell lines with the use of TRIzol reagent and was further purified with the use of an RNeasy Mini kit (QIAGEN, Valencia, CA) according to the manufacturers’ instructions. We then used a high-performance liquid chromatography-purified T7-(dT) primer (5′-GGCCAGTGAATTGTAATACGACTCACTATAGGGAGGCG G-[dT] 24-3′; Affymetrix Inc, Santa Clara, CA) to synthesize double-stranded cDNA from the total RNA; the cDNA was then purified with the use of a phase lock gel (Eppendorf, Westbury, NY), phenol–chloroform extraction, and ethanol precipitation. Biotin-labeled complementary RNA (cRNA) was transcribed in vitro from the cDNA with the use of an ENZO Bioarray High Yield RNA Transcript Labeling kit (Affymetrix Inc), purified with the use of RNeasy columns (QIAGEN), and precipitated with ethanol. The biotin-labeled cRNA was quantified with the use of a spectrophotometer at wavelengths of 260 and 280 nm; fragmented by resuspension in 200 mmol/L Tris-acetate (pH 8.1), 500 mmol/L KOAc, and 150 mmol/L MgOAc; and hybridized to U133A GeneChip microarrays (Affymetrix Inc) according to the manufacturer's instructions. The arrays were washed, stained first with a streptavidin–phycoerythrin conjugate (Molecular Probes, Eugene, OR) and then with biotinylated anti-streptavidin antibody (Vector Laboratories, Burlingame, CA), and scanned to measure signal intensities using a GeneArray Scanner (Affymetrix). Fluorescence intensities were analyzed using Microarray Suite (MAS 5.0) software (Affymetrix). Complete microarray protocols can be found at the Affymetrix website (http://www.affymetrix.com/support/technical/manuals.affx).
Overexpression of STK31
The changes in sensitivity to ABT-737 were assessed following transfection of an empty vector (pJP1563, purchased from DNASU, Arizona State University Biodesign Institute) and a plasmid vector encoding STK31 gene (clone ID: HsCD00039084, purchased from DNASU). RS4;11 cells were cultured as described under cell culture in 35-mm wells. The cells were transiently transfected by electroporation (BioRad) with the pJP1563 expression vector with or without a cDNA insert for STK31. 33 Forty-eight to 96 h after the transfection, the expression of STK31 by RT-PCR, apoptosis by ABT-737 using flow cytometry, and cytotoxicity of ABT-737 by DIMSCAN were assessed in the transfected cells.
Statistical analyses
Differences in gene expression, cytotoxicity, and apoptosis were assessed by unpaired two-sided Student's t tests. Statistical analyses were performed with R for Windows version R-2.4.0. (R Foundation for Statistical Computing, Vienna, Austria) and STATA Release 9 (StataCorp, College Station, TX). P values less than 0.05 were considered statistically significant. All statistical tests were two-sided.
Results
Post-transcriptional caspase-dependent Mcl-1 cleavage in response to ABT-737 in ALL cell lines
We previously reported that Mcl-1 cleaves in response to ABT-737 treatment in ABT-737-sensitive ALL cell lines while the cleavage was not observed up to 1 μmol/L in relatively resistant cell lines. 33 Using an anti-human Mcl-1 antibody produced by immunizing animals with a synthetic peptide corresponding to residues surrounding Leu210 of human Mcl-1, we were able to detect the differences in cleavage of Mcl-1 in two ABT-737-resistant and two ABT-737-sensitive cell lines after treating the cells with either 1 μmol/L (resistant cell lines: NALM-6 and CCRF-CEM, Figure 1a, lanes 1–8) or 100 nmol/L (sensitive cell lines: COG-LL-319 and RS4;11, Figure 1a, lanes 9–16 with full length Mcl-1 being completely cleaved in RS4;11) of ABT-737. Because the cleaved Mcl-1 products were prevalent in the ABT-737-sensitive cell lines, we chose to investigate the mechanisms of Mcl-1 cleavage in ALL cells. Two reports showed two different mechanisms of Mcl-1 degradation. 38 First, by ultraviolet (UV) irradiation, Mcl-1 degradation occurred via a proteasome in HeLa cells and neutrophils.39,40 In this case, the activation of Mcl-1 ubiquitin ligase E3 (MULE) or other E3 ligases polyubiquitinated Mcl-1 during apoptosis while USP9X, a deubiquitinase, stabilized Mcl-1.38,41 Other reports showed that Mcl-1 is subject to caspase-dependent cleavage at two distinct sites within the N-terminus during apoptosis. However, both studies assessed the cleavage extracellularly,41,42 bringing into a question whether the cleavage was an artifact of preparation. In the current study, to identify whether Mcl-1 cleavage is via caspases or is proteasomal, we pretreated sensitive (COG-LL-319) and resistant (NALM-6) cell lines with a pan-caspase inhibitor (boc-d-fmk) or a proteasome inhibitor (MG-132) followed by ABT-737 treatment (COG-LL-319: 200 nmol/L, NALM-6: 10 μmol/L, Figure 1b and c). We observed that Mcl-1 was cleaved in NALM-6 when treated with 10 μmol/L (concentration not likely achievable in vivo 43 ) ABT-737 for 6 h (Figure 1b, lane 2). In both NALM-6 (Figure 1b, lanes 3 and 4) and COG-LL-319h cell lines (Figure 1c, lanes 3 and 4), boc-d-fmk completely abrogated the cleavage of Mcl-1, while MG-132 did not affect the cleavage (Figure 1b and c, lanes 7 and 8). In a separate experiment, ABT-737 treatment did not affect MCL1 mRNA in both ABT-737-sensitive (COG-LL-319) and ABT-737-resistant (NALM-6) cell lines, indicating that the changes in Mcl-1 protein in response to ABT-737 are due to post-transcriptional modulation (data not shown). These data demonstrated that the cleavage of Mcl-1 in response to ABT-737 treatment is caspase-dependent.
Identification of caspase cleavage site(s) in Mcl-1 in ALL cells treated with ABT-737
Two different caspase cleavage sites were previously reported, aspartate 127 (D127) or aspartate 157 (D157) within the N-terminus of Mcl-1 (Supplementary Figure 1a). 41 In the current study, a FLAG/myc-tagged Mcl-1 fragment (127–350 amino acids: Mcl-1 Δ127) and FLAG/myc-tagged wild-type Mcl-1 (1–350) were expressed in NALM-6 cells (Supplementary Figure 1c) by plasmid transfection followed by selection with an antibiotic. However, direct transfection of those plasmids failed to sustain expression between the first and the fifth passage of the cells (Supplementary Figure 1d). To enable more sustained expression of Mcl-1 variants, we employed a lentiviral expression system as described under “Materials and methods” section (Figure 2a). After selecting for stably-transduced cells using blasticidin, the expression of Mcl-1 variants in NALM-6 cell line was maintained for the duration of our experiments (Figure 2b). When the NALM-6 cells transduced with lentiviral vectors encoding full-length V5-tagged Mcl-1 (1–350: Figure 2b, lanes 1 and 2) or the D127A (Figure 2b, lanes 3 and 4) variant were treated with ABT-737, immunoblotting showed only the bands consistant with the endogenous Mcl-1 158-350 fragment (endogenous Mcl-1 Δ157) and also the V5-tagged Mcl-1 158-350 fragment (Mcl-1 Δ157-V5, lanes 2, 4). This confirms that D127 is not the cleavage site induced by ABT-737 treatment in NALM-6 cells. Conversely, when the cells transduced with the D157A variant were treated with ABT-737, the band for V5-tagged Mcl-1 Δ157 disappeared, showing that D157 is the site for cleavage by ABT-737 treatment (lanes 5 and 6). The immunoblotting against V5 showed a Mcl-1 Δ127-V5 band and an endogenous Mcl-1 Δ127 in cells transduced with the D157A variant when treated with ABT-737 (lanes 5 and 6, IB: Anti-V5). These data demonstrate that D157 is the Mcl-1 cleavage site in response to ABT-737 (Figure 2b).
The biological role of Mcl-1 fragments is controversial. Some studies reported that the cleavage at D127 and D157 sites not only impaired Mcl-1's anti-apoptotic activity, but converted Mcl-1 to a pro-apoptotic protein.41,44 However, these data have been disputed by other studies42,45 since the previous studies conducted the experiments on the cleavage of Mcl-1 in extracts of HeLa and Jurkat cells. This leaves open the possibility that extracellular proteolytic changes account for the results. To investigate whether ABT-737 cytotoxicity is affected when D157 cleavage of Mcl-1 is inhibited, we introduced the D157A mutation in Mcl-1 and assessed the cytotoxicity of ABT-737 in ALL cells. ABT-737 cytotoxicity was significantly abrogated in cells expressing the Mcl-1 D157A variant (survival fraction: 18.3 ± 4.5%) compared with non-transduced cells, cells transduced with an empty vector control, Mcl-1 full length (1–350), or Mcl-1 D127A (Figure 2c), for which the survival fractions were 7.3 ± 2.9%, 8.0 ± 1.6%, 4.4 ± 2.2%, and 5.2 ± 2.5%, respectively. These data suggest that D157 cleavage of Mcl-1 enhanced ABT-737 cytotoxicity in ALL cells.
Lost interaction between pro-apoptotic Bcl-2 family (Bim) and Mcl-1 Δ157
We then examined the differences in localization within cells between wild-type Mcl-1 (1–350) and the cleaved Mcl-1 (Δ157). NALM-6 cells expressing V5-tagged Mcl-1 1–350 and V5-tagged Mcl-1 Δ157 were cultured and their cytosol and mitochondria were fractionated. The voltage-dependent anion channel (V-DAC) was used as a marker for separation between cytosol and mitochondria. As shown in Figure 3a, the full length Mcl-1 resided in both cytosol and mitochondria with mitochondrial Mcl-1 being more abundant (Figure 3a, lanes 1 and 3). On the other hand, Mcl-1 Δ157 was exclusively present in the mitochondrial fraction, suggesting that after cleavage, the Mcl-1 fragment either translocates to the mitochondria or that Mcl-1 fragments in the cytosol are rapidly degraded in the cytosol, of which the latter is less likely as different rates of degradation have not been reported for Mcl-1 depending on their localization.
Mcl-1 has a large N-terminal region that is not homologous to Bcl-2, and the molecular characteristics of the N-terminal 170 amino acids of Mcl-1 (determining the unique properties of Mcl-1, i.e. tissue expression, specialized roles in its rate of turnover, switching cell fate) are not well known. 46 We studied the differences in the interaction of Mcl-1 variants (wild type vs. Mcl-1 Δ157) with the pro-apoptotic Bcl-2 proteins, Bim and Bak by immunoprecipitation. Since NALM-6 does not express Bak protein, we first expressed V5-tagged Mcl-1 full length (1–350) and Mcl-1 Δ157 in HEK-293FT cells. Then, using the exogenously expressed Mcl-1-V5 protein, immunoprecipitation was performed to detect target proteins (Bim or Bak) that interacted with V5-tagged wild-type Mcl-1 or Mcl-1 Δ157 in the lysate of RS4;11 cells, which is one of the ABT-737-sensitive cell lines. The results showed that only the full length wild-type Mcl-1 interacts with Bim or Bak (Figure 3b) while only minimal interaction was seen between the Mcl-1 Δ157 variant and Bim or Bak. In addition, to investigate whether the Mcl-1 cleavage at D157 affects caspase activation mediated by ABT-737, we treated NALM-6 cells transduced with lentiviral vectors encoding wild-type Mcl-1 (1–350), Mcl-1 D157A, and Mcl-1 Δ157, and then caspases-9 and -3 activation was assessed at 2, 4, and 6 h of ABT-737 treatment by immunoblotting against 37/35 kDa caspase-9 cleaved form and 17/19 kDa caspase-3 cleaved form that represent the activation of the enzymes. From 4 h of ABT-737 treatment, the activation of both caspases-9 and -3 was greater in cells transduced with Mcl-1 Δ157 vector relative to cells expressing wild-type Mcl-1 (1–350) or the Mcl-1 D157A variant (Figure 3c). These data suggest that the anti-apoptotic property of Mcl-1 is abolished once the D157 site is cleaved due to the loss of interaction between Mcl-1 and members of the pro-apoptotic Bcl-2 family of proteins, such as Bim.
ABT-737-induced apoptosis in ALL cells with mutated phosphorylation sites
There are six serine-threonine phosphorylation sites in Mcl-1,
47
and the phosphorylation of those sites, especially near cleavage sites, are reported to affect the stability of Mcl-1.
47
However, some studies showed conflicting results that might be due to the differences in models and the experimental systems that were used for those studies.41,42,44,45 In human ALL cells, we investigated the effect of phosphorylation site on the Mcl-1 cleavage as well as on the cytotoxicity of ABT-737 by expressing Mcl-1 that was mutated at two phosphorylation sites (S159A and T163A) near the D157 cleavage site. Both Mcl-1S159A- and Mcl-1T163A-expressing NALM-6 cells showed reduced Mcl-1 cleavage and caspase-3 cleavage (Figure 4a left), and caspase-3 enzymatic activation was significantly lower in both Mcl-1S159A—(P = 0.016) and Mcl-1T163A—(P < 0.009) expressing NALM-6 cells compared with Mcl-11-350 cells after 4 h of incubation with ABT-737 (Figure 4a right). In addition, ABT-737 was significantly less cytotoxic in cells with mutated Mcl-1 relative to Mcl-11-350-transduced cell (99 ± 9.7% and 104 ± 7.8% vs. 67 ± 6.7%; P < 0.01, Figure 4b). To determine whether apoptosis contributed to the differences in cytotoxicity among the NALM-6 cells transduced with different Mcl-1 variants, we compared Annexin-V positivity in reponse to ABT-737 treatment. Fewer apoptotic cells were detected in Mcl-1S159A (12.3 ± 1.3%, P = 0.001) and Mcl-1T163A (13.7 ± 2.6%, P = 0.002) compared with Mcl-11-350 (24.6 ± 0.4%) after 24 h of incubation with ABT-737 2 μmol/L (Figure 4c).
Mcl-1 cleavage by ABT-737 and cytotoxicity of ABT-737 in NALM-6 cells transduced with Mcl-1 variants. (a) Reduced Mcl-1 cleavage (left) and caspase-3 cleavage (left) and caspase-3 activity (right) in Mcl-1S157A and Mcl-1T163A relative to Mcl-11-350. Left: the cells were treated with ABT-737 10 μmol/L for 0, 2, and 4 h. The Mcl-1 cleavage was monitored by immunoblotting against V5 and apoptosis by immunoblotting against caspase-3. β-Actin was used as a loading control. The data are representative of two independent experiments. Right: the cells were treated with ABT-737 10 μmol/L for 0 and 4 h. Caspase-3 enzyme activity was assessed by a fluorescent assay kit as described in “Materials and methods” section. Each conditions include three biological replicates that consists of three technical replicates (n = 9). *P < 0.05; **P < 0.001 relative to Mcl-11-350. (b) Mcl-1S157A, Mcl-1T163A, and Mcl-11-350 cells were seeded in 96 plates 16 h before drug treatment. The cells were treated with ABT-737 2 μmol/L for 24 h, and the survival was assessed using DIMSCAN as described in “Materials and methods” section. Fractional survival (%) was calculated relative to the survival of cells treated with vehicle control. Each condition had 12 replicates and the results were reproducible in a repeat experiment. (c) Effect of mutations in phosphorylation sites on apoptosis induced by ABT-737. The cells with Mcl-1 variants were seeded in six-well plates 16 h before treatment. The next day, the cells were treated with ABT-737 2 mmol/L for 24 h. After harvesting, the cells were stained with antibody against Annexin-V labeled with FITC. Fifteen minutes prior to analyzing samples, propidium iodide (PI) was added to cells for counterstaining. Then, the samples were analyzed for annexin-V/PI positivity using flow cytometry. The results of three replicates for each condition are summarized. Closed bars: the percentage of apoptosis in cells treated with vehicle control; open bars: the percentage of apoptosis in cells treated with ABT-737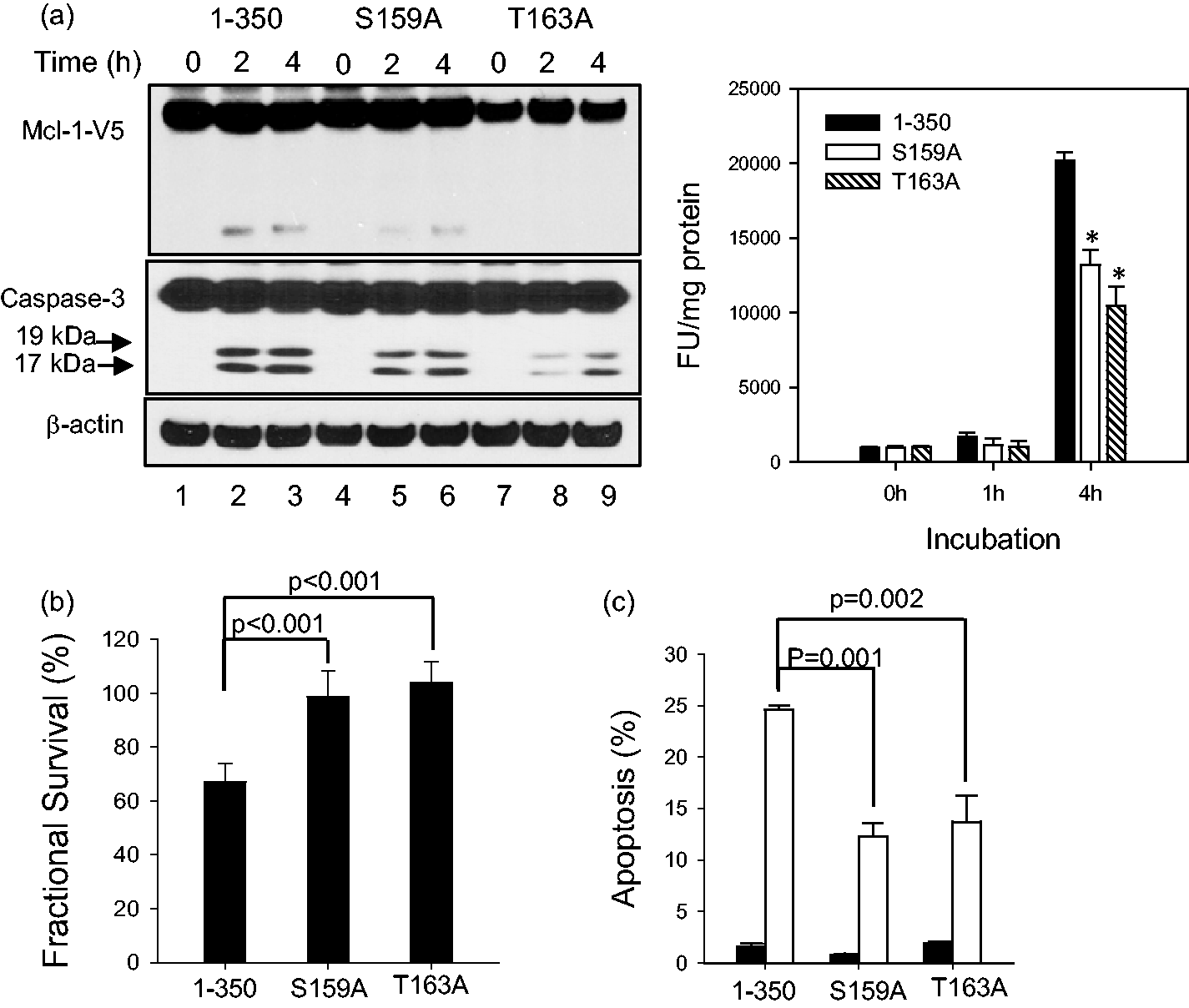
Serine-threonine kinases are associated with ABT-737-resistance
To identify mechanisms that confer drug resistance to ABT-737 in ALL cell lines, we utilized Affymetrix U133A GeneChips to compare the genome-wide expression profiles in two sets of samples: (1) ALL cell lines with various sensitivity to ABT-737 collected at their log-growth phase; (2) two ALL cell lines, COG-LL-319 and RS4;11 that are constitutively sensitive to ABT-737, but selected for resistance to ABT-737 by step-wise selection. This experimental design allowed us to determine whether there is a common mechanism of resistance to ABT-737 between constitutive and developed resistance. We identified 32 genes whose expression is significantly correlated with IC90 values (P < 0.001) (Figure 5a). Of the 32 genes identified, we investigated the role of STK31, serine-threonine kinase 31 on ABT-737 sensitivity as all Mcl-1 phosphorylation sites are serine or threonine. We transiently overexpressed STK31 by transient transfection as described in “Materials and methods” section in the most sensitive cell line to ABT-737, RS4;11. STK31 expression was significantly higher in the cells transfected with STK31 relative to the cells transfected with the empty vector (Figure 5b, P < 0.001), and the cells overexpressing STK31 were significantly more resistant to ABT-737 compared to an empty vector control (Figure 5c, P = 0.003). However, STK31 was not significantly different between constitutively sensitive cells and the cells selected for resistance to ABT-737. In fact, none of the genes were associated with both constitutive resistance and developed resistance to the drug (Figure 5a), indicating that the mechanisms of constitutive resistance are independent of selected (by step-wise selection) resistance in ALL cells.
Serine/threonine kinase 31 (STK31) on ABT-737 sensitivity and Mcl-1 stability. (a) Hierarchical clustering heat map of gene expression. Expression matrix displays 32 different probe sets that were correlated with IC90 values of ABT-737 in nine ALL cell lines as well as in two cell lines (RS4;11 and COG-LL-319) selected for resistance to ABT-737 using step-wise selection (P < 0.001). mRNAs of COG-LL-319 and RS4;11 cells were isolated when cells became resistant to ABT-737 at 200 nmol/L (COG-LL-319 200 and RS4;11 200), 400 nmol/L (COG-LL-319 400 and RS4;11 400), and 800 nmol/L (COG-LL-319 800 and RS4;11 800). Genes are ranked according to its correlation to IC90 (positive to negative) expression data from resistant selected COG-LL-319 and RS4;11 cells were not used for correlation analysis, but included in heatmap. With few exceptions, the resistant gene expression pattern from 9 ALL cells is different from those of COG-LL-319 and RS4;11. The expression of FGD6 and HABP2 is higher in all resistant cells. The data are presented in a matrix format in which each row represents an individual gene and each column represents a tissue sample. Each cell in the matrix represents the expression level of a gene feature in an individual tissue sample. The red and green coloring in the cells reflects relatively high and low expression levels, respectively. The expression of each gene in each sample was normalized in the pseudocolored expression matrix based on the number of standard deviations above (red) and below (blue) the median expression value (black) across all samples. Samples and genes were optimally clustered and ordered using Pearson correlation distance metric with complete linkage distance measurements. (b) STK31 overexpression effect on ABT-737 sensitivity. STK31 construct was transfected into the RS4;11 cell line, and the fold increase in the transcription of STK31 in cells transfected with STK31 relative to ones with empty vector is shown (top left, n = 3, ***P < 0.01). STK31 expression was measured by real-time RT-PCR. (c) The changes in cytotoxicity in response to ABT-737 treatment at 3 and 10 nmol/L at 72 h of incubation were measured (cytotoxicity using DIMSCAN, n = 6, *P < 0.05)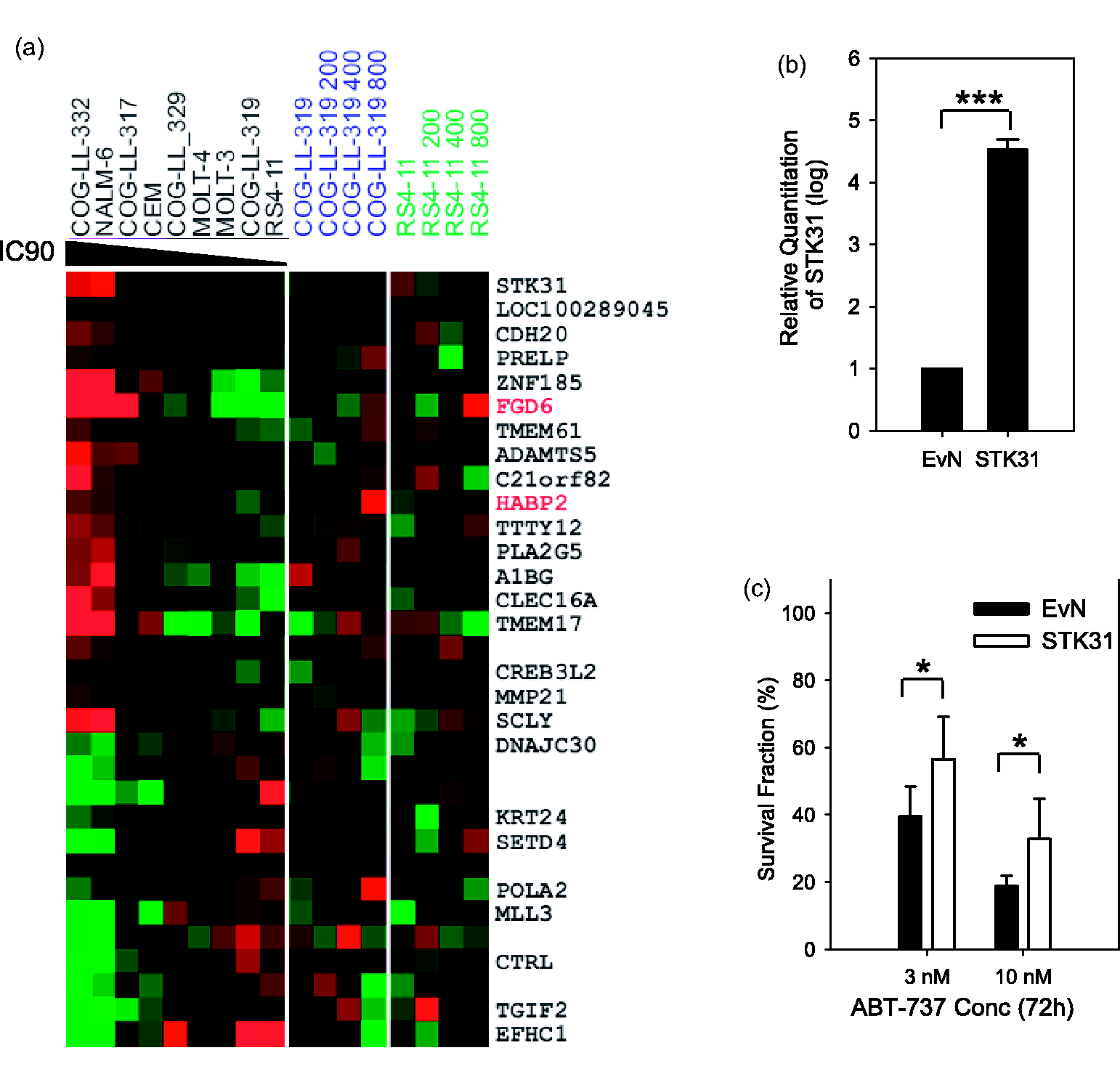
PI3K inhibitors (PI3Ki) are reported to enhance the anti-tumor activity of ABT-737.
48
GSK-3 regulates Mcl-1 stability via PI3K/Akt, and thus affects apoptosis in FL5.12, a murine pro-B-cell lymphoid cell line.
49
GSK-3 has been reported to affect phosphorylation of S155, S159, and T163 residues in Mcl-1. We explored whether the responses to the addition of a PI3Ki (LY294002) to ABT-737 differed among Mcl-1 variants and whether the effect of GSK inhibition (CT99021) was compromised in cells with mutations in phosphorylation sites of Mcl-1. In Mcl-11-350, Mcl-1 cleavage at D157 was clear after 2 h of incubation with ABT-737 (10 μmol/L), and the addition of LY294002 (8 μmol/L), which is reported to reduce Mcl-1 protein by ubiquitinylating in HeLa cells,
49
resulted in greater Mcl-1 cleavage while the Mcl-1 cleavage appears to be minimal in Mcl-1S159A and Mcl-1T163A by ABT-737 + LY294002 combination (Figure 6a). In Mcl-1S159A and Mcl-1T163A, cleaved caspases 3 (17 kDa and 19 kDa) were less than those seen in Mcl-11-350 after the cells were treated with ABT-737 + LY294002 (Figure 6a). The Mcl-1 cleavage by ABT-737 + LY294002 was diminished by the addition of CT99021 (2 μmol/L) in Mcl-11-350 (Figure 6b). Our data show that phosphonegative mutations of S159 and T163 both stabilize Mcl-1, following ABT-737 treatment.
PI3K inhibitor effect on Mcl-1 cleavage and cytotoxicity induced by ABT-737. (a) Enhanced ABT-737-induced Mcl-1 cleavage and caspase-3 activation by PI3K inhibitor (PI3Ki). NALM-6 cells (Mcl-11-350, Mcl-1S157A, or Mcl-1T163A) were incubated overnight in a humidified incubator as described in “Materials and methods” section. Then, the cells were treated with vehicle (0.1% DMSO) or pretreated with LY294002 at 8 μmol/L for 2 h followed by ABT-737 at 10 μmol/L for 1, 2, or 4 h. Then, the cells were harvested and the whole cell lysates were subjected to immunoblotting against V5 and caspase-3. β-Actin was used as a loading control. The passage numbers of the cells after selection with blasticidin and the time in culture before experiments were the same for Mcl-11-350, Mcl-1S157A, and Mcl-1T163A. (b and c) GSK inhibition effect on PI3K inhibitor (PI3Ki, LY294002, LY) + ABT-737 combination effect. The cells were treated with vehicle, a GSK inhibitor (GSKi, CT99021, CT), or a GSKi + a PI3Ki for 2 h followed by ABT-737 (a) 10 μmol/L for 2 h for immunoblotting assay (b) and 24 h for cytotoxicity assay (c). Then, the cells were lysed and the protein samples were subjected to immunoblotting against V5. β-Actin was used as a loading control (b). The survival of the cells was assessed by a DIMSCAN system. All percent fractional survival values were normalized to control. Each bar represents the mean value for 12 replicates and error bars correspond to standard deviations. Experiments were performed twice and were consistently repeatable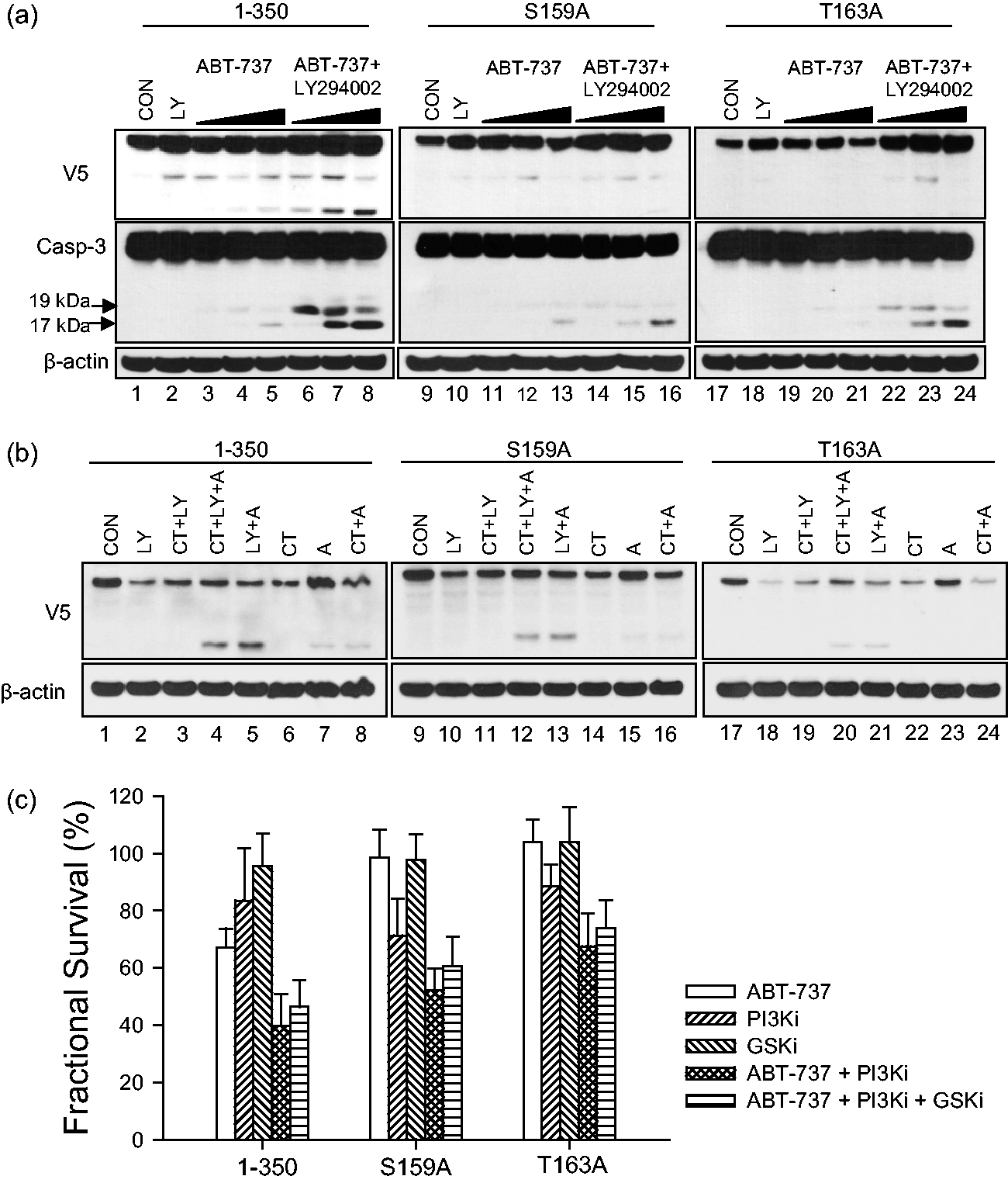
These results demonstrate that apoptotic activity of ABT-737 was reduced in cells with Mcl-1S159A and Mcl-1T163A variants. The differences in cell death were assayed using DIMSCAN (Figure 6c). As anticipated, addition of a PI3Ki significantly enhanced ABT-737 cytotoxicity in wild type and both Mcl-1 variants compared with ABT-737 alone (Figure 6c, P < 0.01). However, unlike the previous report 49 the reversal effect of GSKi on the cytotoxicity of ABT-737 + PI3Ki was not significant in all three models (Figure 6c; P = 0.06–0.28).
Discussion
In this study, we have shown that post-transcriptional regulation of Mcl-1 occurs in response to ABT-737 treatment in ALL cell lines and that such regulation differs between ALL cell lines sensitive and resistant to ABT-737. Unlike the other anti-apoptotic Bcl-2 family of proteins, Mcl-1 contains three putative BH domains and a large N-terminal region that is rich in regulatory motifs. 46 The N-terminal residues 1–170 has three ubiquitination sites, two caspase cleavage sites, and six phosphorylation sites (Supplementary Figure 1a), which affect the degradation, localization, and phosphorylation of the protein. 47 The degradation of Mcl-1 is attributed to proteasomal degradation and caspase-dependent cleavage.38,41 Although molecular characteristics of Mcl-1 after cleavage are controversial,41,45 it appears that cleavage of Mcl-1 is an important process in determining the fate of ALL cells. Our data are the first to demonstrate that Mcl-1 is cleaved at D157, but not D127, by caspases after ABT-737 treatment by expressing Mcl-1 protein variants in leukemia cells, which also showed that Mcl-1 is post-translationally modulated after ABT-737 treatment. The concentrations of ABT-737 required to cleave Mcl-1 were drastically different between resistant cell lines (10 μmol/L) and sensitive cell lines (100 nmol/L), suggesting that Mcl-1 in resistant cells will not be cleaved after ABT-737 treatment at clinically achievable concentrations.
Subsequent experiments revealed that Δ157 fragment does not interact with the pro-apoptotic Bcl-2 family of proteins, indicating that the anti-apoptotic property of Mcl-1 that inhibits pro-apoptotic Bcl-2 family of proteins by forming heterodimers is lost in Mcl-1 Δ157 fragment. Additional evidences show that Mcl-1 Δ157 may contribute to ABT-737 cytotoxicity by enhancing caspase activation. However, it does not appear that the Mcl-1 Δ127 variant affects the cytotoxicity of ABT-737 when expressed in ALL cells. Considering that the endogenous Mcl-1 protein is more abundant than exogenously expressed Mcl-1-V5 in these experiments, the biological consequences of the Mcl-1 fragment Δ157 on cell viability after ABT-737 treatment might have been greater than was observed in our experiments. An important aspect of these data is that our experiments used exogenously expressed Mcl-1 and Mcl-1 fragments within ALL cells, and the subsequent experiments were conducted using living cells while the majority of the published data have utilized cell extracts or non-human cells due to the technical challenges in expressing proteins in suspension cells.50–53
As we confirmed that D157 is more susceptible to ABT-737-induced cleavage compared to D127, we investigated the role of phosphorylation near the cleavage site (S159 and T163) on Mcl-1 cleavage. It has been reported that initial priming phosphorylation of T163 is required for S159 phosphorylation, 54 which is a characteristic of other GSK-3 targets. 55 In addition, GSK-3 has been identified as the kinase targeting S159 residue by multiple groups, and phosphorylation of this residue led to significant decreases in Mcl-1 protein expression.49,54,56 The role of adjacent phosphorylation sites on Mcl-1 stability was investigated by mutating S159 and T163 in the current study. Our data demonstrated that Mcl-1 cleavage is substantially reduced in response to ABT-737 treatment in cells with mutated phosphorylation sites (Figure 4). In addition, cytotoxicity via inducing apoptosis by ABT-737 is also significantly diminished in cells expressing Mcl-1S159A and Mcl-1T163A compared with Mcl-11-350, suggesting that Mcl-1 cleavage requires phosphorylation of S159 and/or T163. It has been suggested that phosphorylation of Mcl-1 by GSK-3 promotes its accelerated degradation. 49 The inhibition of GSK-3 using CT99021 did not affect the cleavage of Mcl-1 to Mcl-1 Δ157 which was increased in treatment using the combination of ABT-737 + PI3Ki (Figure 6b). However, the GSK-3 inhibition did not significantly affect the combination cytotoxicity of ABT-737 + PI3Ki, even in cells with mutated T163, a GSK-3 priming site (Figure 6c). Our data suggest that the role of GSK-3 in cytotoxicity induced by the combination of ABT-737 and PI3Ki is not crucial.
While constitutive levels of Mcl-1 was proposed as a molecular marker for resistance for ABT-737 in several types of tumors,57,58 we previously reported that constitutive expression of Mcl-1 itself may not be the determining factor for the resistance to ABT-737 in ALL models. 33 In the current study, we demonstrated that Mcl-1 is cleaved at D157 site after ABT-737 treatment in ALL cells and the cleavage results in the generation of pro-apoptotic fragment of Mcl-1 Δ157. As ABT-737 has low binding affinity to Mcl-1, combining a drug that regulates Mcl-1 has been suggested as a strategy for the drug development. Our data suggest that drugs activating capases could potentially overcome resistance to ABT-737, at least in ALL cell lines. In addition, the phosphorylation sites need to be studied for the effect of serine-threonine phosphorylation on the cleavage and stability of Mcl-1.
In conclusion, our study is first to show (1) the intracellular expression of Mcl-1 protein variants in ALL cells, (2) regulation of Mcl-1 protein in response to ABT-737 treatment, and (3) that cleavage of Mcl-1 in ALL cells plays a role in cytoxicity. These data indicate that future studies on serine-threonine kinases in association with Mcl-1 phosphorylation at S159 and T163 sites that affect the stability of the Mcl-1 protein may potentially identify a marker for sensitivity and resistance to ABT-737.
Footnotes
Author's contributions
All authors participated in the design, interpretation of the studies and analysis of the data and review of the manuscript; RY, CPH, and MHK conducted the experiments, MHK and CPR wrote the manuscript.
ACKNOWLEDGEMENTS
The authors thank Abbott Laboratories for the supply of ABT-737. We also thank Children's Oncology Group (COG) for providing ALL cell lines. The drug supplier had no role in design of the study; the collection, analysis, or interpretation of the data; the writing of the manuscript; or the decision to submit the manuscript for publication. This study was supported by the Cancer Prevention and Research Institute of Texas (Grant RP101042). Microarray analyses were performed in Dr Timothy Triche's laboratory at University of Southern California, and the data analysis was conducted by Dr Ju-Seog Lee at MD Anderson Cancer Center. This study was supported by the Cancer Prevention and Research Institute of Texas (Grant RP101042).