
Abstract
Subepithelial fibrosis is one of the common pathological features of asthmatic airway remodeling. During subepithelial fibrosis, type I collagen becomes the most abundant extracellular protein component. Studies have shown that Notch signaling participates in the progression of fibrosis; however, whether Notch signaling is involved in regulating type I collagen expression in airway fibroblasts remains unclear. The aim of the present study was to examine whether Notch signaling can regulate type I collagen expression in airway fibroblasts and to explore the underlying molecular mechanisms. Here, the expression of Notch signaling components was examined in mouse L929 cells and human MRC-5 cells. After upregulating or downregulating Notch signaling in these cell lines, col1α1 and col1α2 expression was examined. Using gene reporter assays, site-directed mutagenesis, and ChIP assays, the role of Hes1 binding sites in both the mouse and human COL1A1 and COL1A2 promoters was investigated. This study revealed that Notch signaling-related molecules (including Notch1, Hes1, and others) are expressed in L929 and MRC-5 cells and that Notch signaling regulates the expression of col1α1 and col1α2 in both cell lines. Additionally, over-expression of the Notch intracellular domain resulted in activation of the COL1A1 and COL1A2 promoters, and site-directed mutagenesis reporter assays revealed that Hes1 proteins might augment both mouse and human COL1A1 and COL1A2 promoter activity. Furthermore, ChIP assays confirmed that Hes1 binds to the COL1A1 and COL1A2 promoters in both L929 and MRC-5 cells. Therefore, it is reasonable to assume that Notch signaling can directly upregulate COL1A1 and COL1A2 promoter activity through a Hes1-dependent mechanism, which could serve as a possible target for pharmacotherapy of airway subepithelial fibrosis.
Introduction
The most typical pathological feature of airway remodeling in asthma is subepithelial fibrosis,1,2 which is caused by excessive accumulation of fibroblasts and deposition of extracellular matrix. The major component of extracellular matrix is type I collagen. Type I collagen is a heterotrimer of two alpha1 (col1α1) and one alpha2 collagen (col1α2) subunits. In fibroblasts or myofibroblasts, the two collagen polypeptides are encoded by the type I collagen A1 and A2 genes (COL1A1 and COL1A2, respectively). 3 Transforming growth factor-β1 (TGF-β1) is a good inducer for collagen synthesis of fibroblasts, which promotes subepithelial fibrosis of asthma.4,5 Though current asthma therapies, such as inhaled corticosteroids, β2-agonists, and M cholinergic receptor antagonists are effective in treating acute attacks or relieving symptoms, 6 there is currently no effective therapy for subepithelial fibrosis in asthma.
Notch signaling is highly conserved throughout evolution. This pathway mediates signaling between neighboring cells during metazoan development via Notch receptors (Notch1, 2, 3, and 4), ligands (Jagged1 and 2; Delta1, 3, and 4), and downstream signaling transduction molecules (Hes and Hey).
7
The Notch receptors and ligands are single-pass transmembrane proteins expressed on the cell surface. By binding to the ligands, Notch receptors are activated and trigger sequential release of the Notch intracellular domain (NICD).
8
γ-secretase makes the cytoplasmic cleavage of NICD. N-3,5-difluorophenyl acetyl-
In this study, we examined the expression of Notch signaling-related molecules in lung fibroblasts, observed the regulatory role of Notch in col1α1 and col1α2 expression, and investigated the underlying molecular mechanisms of this pathway.
Materials and methods
Cell culture
The 293 T human embryonic kidney cell line was purchased from American Type Culture Collection, USA (ATCC-CR-3216) and cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (FBS). The mouse fibroblast cell line L929 (GNM28) and human lung fibroblast cell line MRC-5 (GNHu41) were purchased from the Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China. L929 cells were cultured in Roswell Park Memorial Institute 1640 (RPMI-1640) medium supplemented with 10% FBS. MRC-5 cells were cultured in minimum essential medium containing 10% FBS.
Quantitative reserve transcription-polymerase chain reaction (PCR)
Two micrograms of total RNA was reverse transcribed with reverse transcriptase (Promega Corporation, Madison, WI, USA) according to the manufacturer’s instructions. All PCR experiments were performed with Taq polymerase (Promega Corporation, Madison, WI, USA). The following mouse primers were used: Notch1, forward 5′-CGGTGAACAATGTGGATG-3′ and reverse 5′-TGTGCTGACAGAGG-3′; Hes1, forward 5′-AGCACAGAAAGTCATCAAAG-3′ and reverse 5′-TGGCTTAGACTTTCATTTATTC-3′; mouse β-actin (used as an internal control), forward 5′-GGCTGTATTCCCCTCCATCG-3′ and reverse 5′-CCAGTTGGTAACAATGCCATGT-3′. The following human primers were also used: Notch1, forward 5′-GGACCTCATCAACTCACA-3′ and reverse 5′-GTCTCCTCCCTGTTGTTC-3′; Hes1, forward 5′-GCACAGAAAGTCATCAAA-3′ and reverse 5′-GTGCTTCACTGTCATTTC-3′; human β-actin (used as an internal normalized control), forward 5′-CATGTACGTTGCTATCCAGGC-3′ and reverse 5′-CTCCTTAATGTCACGCACGAT-3′.
Western blotting
Proteins were separated by sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. The membranes were saturated for 1 h at room temperature in tris-buffered saline and tween20 (TBST) supplemented with 5% skim milk and immunoblotted overnight at 4℃ with anti-Notch1 (#4380S, CST, Boston, USA), anti-Hes1 (SC-13844, Santa Cruz Biotechnology, California, USA), anti-β-actin antibody (sc-130656; Santa Cruz Biotechnology). The primary antibodies were all recommended for mouse, rat, and human samples. Following incubation with primary antibodies, the membranes were washed, probed with horseradish peroxidase-conjugated goat anti-mouse IgG (SC-2005, Santa Cruz Biotechnology, California, USA) or donkey anti-goat (A0181, Beyotime, Shanghai, China) polyclonal antibody, depending on the species of the primary antibody, and imaged using enhanced chemiluminescence (Amersham Biosciences, Uppsala, Sweden).
Over-expression NICD lentiviral vector infection, TGF-β1, and GSI treatments
Lentiviral constructs expressing NICD were purchased from Sunbio, Shanghai, China. L929 and MRC-5 cells were infected with the lentiviral vectors according to the manufacturer’s instructions. Steady over-expression of NICD in L929 and MRC-5 was verified by PCR or Western blotting as described earlier. Cells were then cultured in 24-well plates for 4 days, supplemented with 5 ng/mL TGF-β1 (H8541, Sigma Aldrich, Shanghai, China) or control solution. On day 5, DAPT (D5942, Sigma Aldrich, Shanghai, China) a classical pharmacological inhibitor of Notch signaling was added at 2 µM.
16
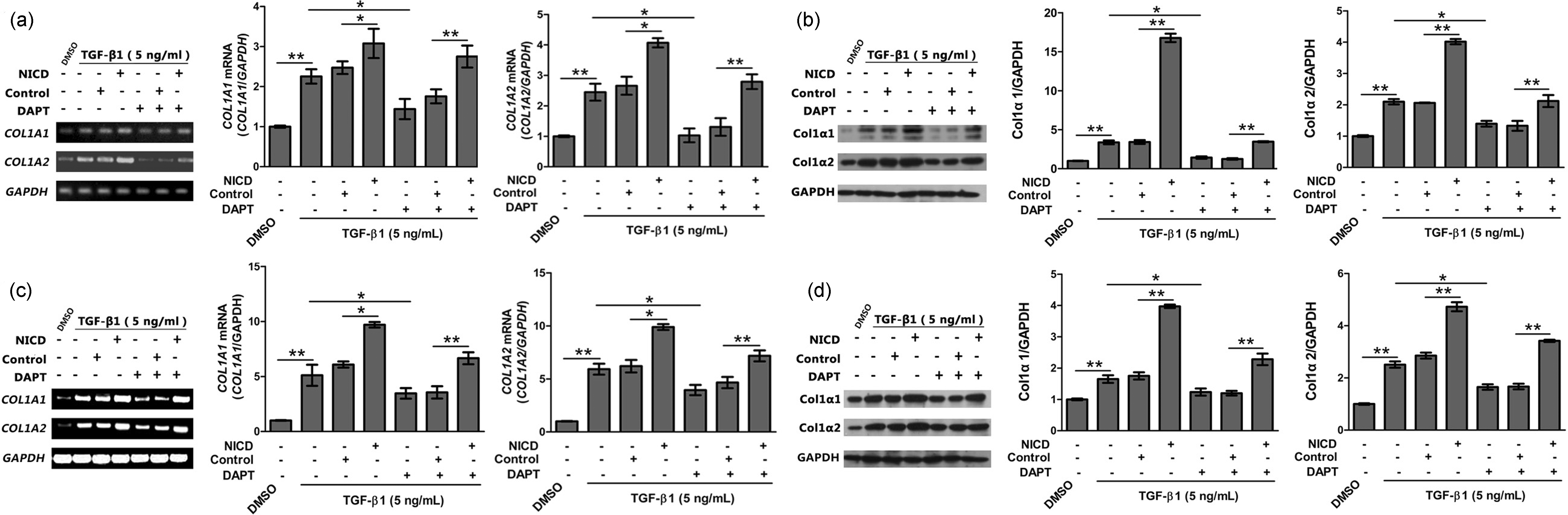
The expression of col1α1 and col1α2 was detected by RT-PCR and western blotting after 48 h (Figure 2).
The expression of Notch1 and Hes1 in mouse and human fibroblasts. (a, b) Notch1 and Hes1 proteins were detected by western blotting in L929 and MRC-5 cells with β-actin as a control for protein loading (n=5). (c, d) Notch1 mRNA and Hes1 mRNA were detected by PCR in L929 and MRC-5 cells with β-actin as a control (n = 5) Over-expression of NICD and treatment with GSI regulates Col1α1 and Col1α2 expression. Mouse L929 and human MRC-5 cells were transfected with lentiviruses that express NICD and were then cultured in 24-well plates for 4 days, treated with 5 ng/mL TGF-β1 or control solution. On day 5, 2 µM DAPT or control solution was added. On day 7, the expression of Col1α1 and Col1α2 was detected by RT-PCR and western blotting using GAPDH as a control. (a) Electrophoretic band and relative expression of COL1A1 and COL1A2 mRNA in L929 (n = 3). (b) Immunoblot and relative intensity of Col1α1 and Col1α2 protein expression in L929 (n = 3). (c) Electrophoretic band and relative expression of COL1A1 and COL1A2 mRNA in MRC-5 (n = 3). (d) Immunoblot and relative intensity of Col1α1 and Col1α2 protein expression in MRC-5 (n = 3). NICD: cells over-expressing Notch intracellular domain, DAPT: γ-secretase inhibitor (GSI), N-3,5-difluorophenyl acetyl-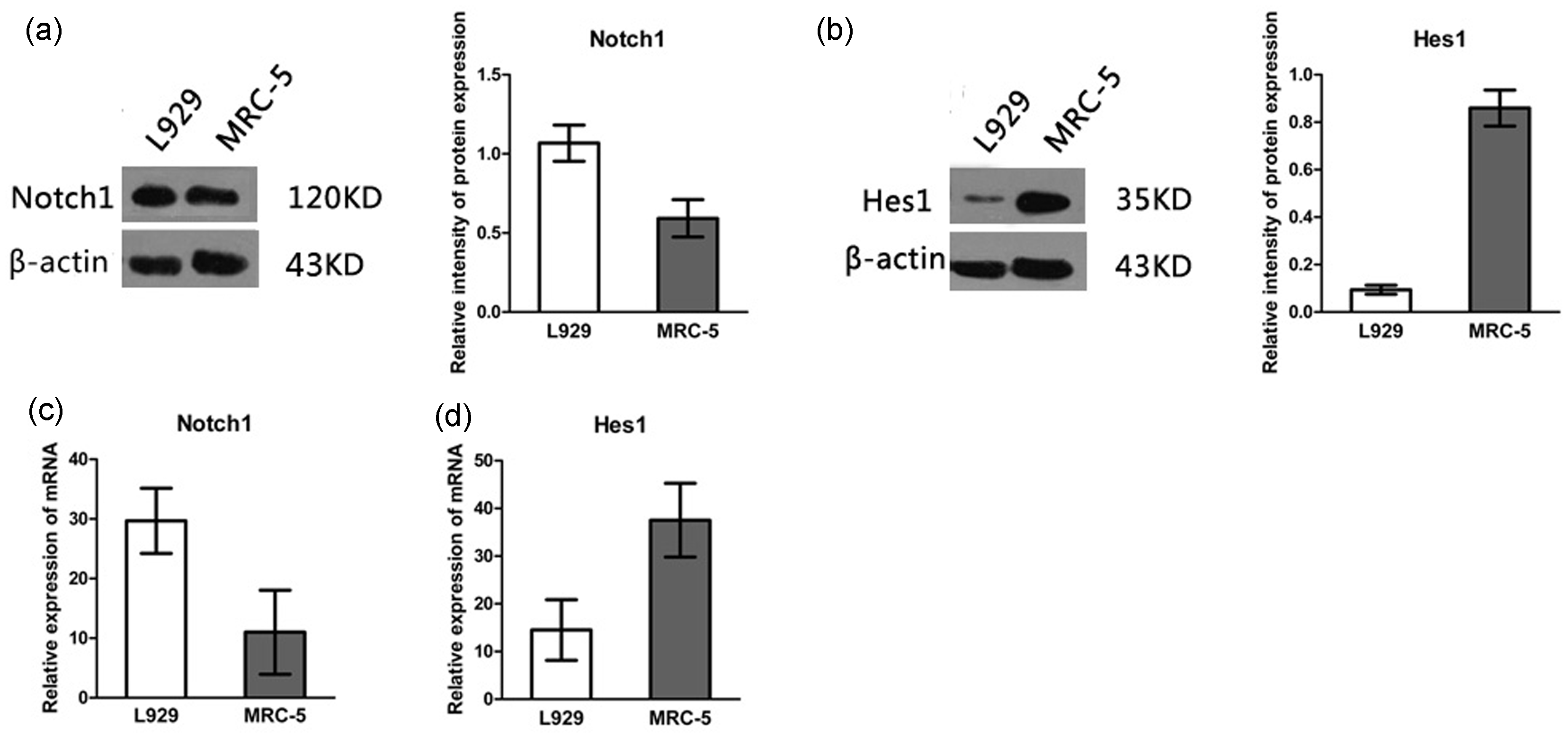
Reserve transcription-PCR (RT-PCR) analysis
RT-PCR was performed on L929 and MRC-5 cells as described in our previous study. 17 The following mouse primers were used: Col1α1, forward 5′-CAAGGTGACAGAGGCATA-3′ and reverse 5′-CAGGAGAACCAGGAGAAC-3′; Col1α2, forward 5′-CTGGGAAACATGGAAACC-3′ and reverse 5′-CCTTGAGGACCACTAGGA-3′; The following human primers were used: Col1α1, forward 5′-AGCAGATCGAGAACATCC-3′ and reverse 5′-CCAGTACTCTCCACTCTTC-3′; Col1α2, forward 5′-GATGGAAACAAGGGTGAA-3′ and reverse 5′-GTCCACTAGGACCAGATG-3′; GAPDH (used as an internal normalized control), forward: 5′-TGGCGCTGAGTACGTCGTG-3′ and reverse 5′-ATGGCATGGACTGTGGTCAT-3′.
Cloning and vector construction
The mouse COL1A1 promoter (from −2301 to +99) was amplified by PCR using primers 5′-CCGAGCTCTTACGCGTGCCTTCAACTCCTGGGG-3′ and 5′-GATCGCAGATCTCGAGGTCTAGACCCTAGACATGTAGACTC-3′. The mouse COL1A2 promoter (from −2000 to +126) was amplified by PCR using primers 5′-CCGAGCTCTTACGCGTTAAATCTGTTTTCTTACCTACCC-3′ and 5′-GATCGCAGATCTCGAGGCAGACTCCTTGTGTTGC-3′. The human COL1A1 promoter (from −2074 to +126) was amplified by PCR using primers 5′-CCGAGCTCTTACGCGTGCCCTGCGGCACCCTGC-3′ and 5′-GATCGCAGATCTCGAGGTCTAGACCCTAGACATGTAGACTC-3′. The human COL1A2 promoter (from −1772 to +228) was amplified by PCR using primers 5′-CCGAGCTCTTACGCGTGACAGGCCTTCCTCCTCTC-3′ and 5′-GATCGCAGATCTCGAGGGCTGAGAAGCCGAAACGC-3′. The amplified promoter fragments were cloned into the pGL3-basic vector (E1751, Promega Life Science, Madison, WI, USA) to generate the pGL3-mCOL1A1, pGL3-mCOL1A2, pGL3-hCOL1A1, and pGL3-hCOL1A2 plasmids. Site-directed mutagenesis of N-boxes in the promoters was performed using the QuickChange kit (210518, Agilent Stratagene, Shanghai, China) according to the manufacturer’s instructions.
Luciferase reporter assays
NICD plasmids (pEF-Bos-NICD) and control plasmids (pEF-Bos) were a kind gift of Prof. Hua Han (the Fourth Military Medical University, Xi’an, China). 293 T cells were transfected with 0.4 µg reporter construct as above, together with increasing amounts (0, 0.05, 0.1, and 0.2 µg) of pEF-Bos-NICD, and 0.04 µg renilla luciferase vector (phRL-TK; Promega, Madison, WI, USA) using Lipofectamine 2000™ (11668, Invitrogen, California, USA). The total amount of transfected DNA was balanced with pEF-Bos. Luciferase activity was assessed 48 h after transfection using a Luminoskan Ascent (Labsystems, Helsinki, Finland, Tibrug, The Netherlands) and a Dual-Luciferase Reporter Assay Kit (E1910, Promega, Madison, WI, USA) according to the manufacturer’s protocol. All luciferase activity was normalized to renilla luciferase activity.
Chromatin immunoprecipitation (ChIP)
ChIP was performed as described previously. 18 PCR primers for ChIP assays included: mouse COL1A1, forward: 5′-CCCTACAGGCACTGGCATT-3′, reverse 5′-ACTGCAGGGGCTATAATTAAAGG-3′; mouse COL1A2: forward: 5′-GAACGGTCCACGATTGCAT-3′, reverse: 5′-TTTCCCTCTGTTGAGATAAGGGTG-3′. human COL1A1: forward: 5′-AACTGCCATCTCAGCACCTC-3′, reverse 5′-TGCAGGGGCTATAATTAAAGGGAA-3′, human COL1A2, forward: 5′-CCGCCGCCGAGGTTCC-3′, reverse: 5′-TGAGAATCCACCGCCCCCC-3′.
Statistical analysis
Statistical analyses were performed using the SPSS19.0 program. Comparisons between groups were analyzed with one-way analysis of variance (ANOVA). Data were expressed as the mean ± standard deviation, and differences with a P < 0.05 were considered statistically significant.
Results
Expression of Notch signaling receptors and downstream molecules in mouse and human fibroblasts
We examined the expression of Notch signaling-related molecules in pulmonary fibroblasts by immunoblot. The results showed that Notch1 was expressed in a murine fibroblast cell line (L929) and a human lung fibroblast cell line (MRC-5) (Figure 1(a) and (c)). However, expression of Notch2, Notch3, and Notch4 was not detected (data not shown). Hes1, a key downstream molecule of Notch signaling, was also detected in both cell lines (Figure 1(b) and (d)). Consistent with the immunoblot results (Figure 1(a) and (b)), real-time PCR assays (Figure 1(c) and (d)) detected mRNAs encoding the aforementioned Notch related molecules. These results suggest that Notch signaling-related molecules are expressed in fibroblasts.
Ectopic over-expression of NICD and treatment with GSI can regulate Col1α1 and Col1α2 expression
To investigate whether Notch signaling can regulate expression of collagen proteins, we upregulated or downregulated Notch signaling in L929 (Figure 2(a) and (b)) and MRC-5 cells (Figure 2(c) and (d)) and then detected expression of col1α1 and col1α2 by RT-PCR (Figure 2(a) and (c)) and Western blotting (Figure 2(b) and (d)). The results showed that in the presence of TGF-β1, expression of col1α1 and col1α2 significantly increased in NICD over-expressing cells at both the mRNA and protein levels relative to the controls (Figure 2; **, P < 0.01; *, P < 0.05). Similarly, downregulating Notch signaling with DAPT decreased the expression of col1α1 and col1α2 both in human MRC-5 and mouse L929 cells (Figure 2; **, P < 0.01; *, P < 0.05).
Sequence alignment analysis of the mouse, rat, and human COL1A1 and COL1A2 promoters
To determine whether Notch signaling directly regulated the COL1A1 and COL1A2 promoters, we analyzed the genomic sequence of the mouse (COL1A1: NC_000077.5; COL1A2: NC_000072.5), rat (COL1A1: NW_047337.1; COL1A2: NW_047688.2), and human (COL1A1: NC_000017.10; COL1A2: NC_000007.13) COL1A1 and COL1A2 promoters. An alignment of the promoters from these three species is depicted in Figure 3, showing the consensus transcriptional start site and translational start sites of the COL1A1 and COL1A2 promoters (Figure 3(a) and (c)). In the 5′ region of mouse, rat, and human COL1A1 or COL1A2 promoters, we found several Hes consensus binding motifs, which are defined as the N-box (CACNAG) (Figure 3(b) and (d)).
(a, c) Sequence alignment of the human, mouse, and rat COL1A1 and COL1A2 promoters. The TSS (transcription start sites), TLSS (translational start sites), and N-boxes were symbolized. (b, d) Schematic of the 5′ region of the COL1A1 and COL1A2 promoters, indicating potential Hes binding sites. (A color version of this figure is available in the online journal.)
Effects of the Hes1-binding sites on the mouse and human COL1A1 and COL1A2 promoters
To observe whether these putative Hes binding sites can modulate transcriptional activity, we amplified the mouse COL1A1 (−2301/ +99) and COL1A2 (−2000/+126) promoter and constructed reporter plasmids pGL3-mCOL1A1 and pGL3-mCOL1A1. Co-transfection of 293 T cells with the reporters and increasing doses of NICD expression vector gradually increased mouse COL1A1 and COL1A2 promoter activity (Figure 4(a) and (b)).
Effects of Hes-binding sites on COL1A1 and COL1A2 promoter. (a) Reporter assay. 293 T cells were transfected with pGL3-mCOL1A1 (0.4 µg) and gradient doses of pEF-Bos-NICD (0, 0.05, 0.1, 0.2 µg) as well as other plasmids as indicated. The relative luciferase activity (firefly luciferase/renilla luciferase) was analyzed 48 h later. The results were presented as the mean ± SD. *, P < 0.05 **, P < 0.01 (n = 3). (b) Reporter assay. The activity of pGL3-mCOL1A2 was analyzed (n = 3). (c) Reporter assay. The activity of pGL3-Mut-mCOL1A1 was analyzed (n = 3). (d) Reporter assay. The activity of pGL3-Mut-mCOL1A2 was analyzed (n = 3). (e) Reporter assay. The activity of pGL3-hCOL1A1 was analyzed (n = 3). (f) Reporter assay. The activity of pGL3-hCOL1A2 was analyzed (n = 3). (g) Reporter assay. The activity of pGL3-Mut-hCOL1A1 was analyzed (n = 3). (h) Reporter assay. The activity of pGL3-Mut-hCOL1A2 was analyzed (n = 3). (i) ChIP assay. Chromatin preparations from L929 cells was immunoprecipitated using anti-Hes1, and co-precipitated DNA fragments were amplified by primers specific for the N-box fragment of the mouse COL1A1 and COL1A2 promoter, respectively. Normal goat serum was used as a control. (j) ChIP assay. Chromatin preparations were collected from MRC-5 cells and were done as in Figure 4(i). m: mouse; h: human; Mut: mutant; phRL: renilla luciferase vector; pEF-Bos-NICD: plasmid expressing Notch intracellular domain; pGL3-COL1A1/COL1A2: COL1A1/COL1A2 promoter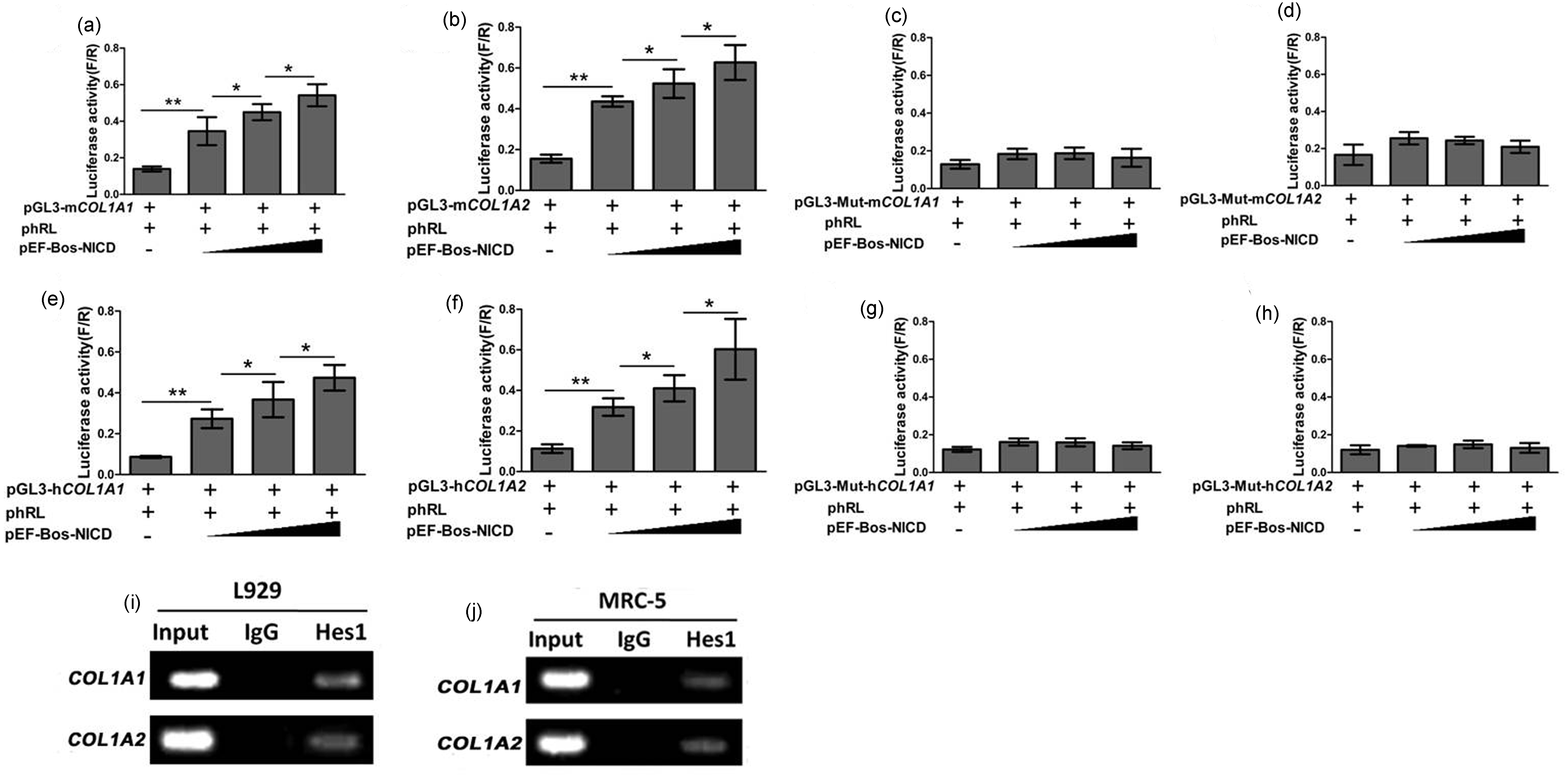
To investigate the function of the N-boxes in the COL1A1 and COL1A2 promoters, we disrupted them by site-directed mutagenesis and examined their activities using the reporter assay as described earlier. As shown in Figure 4(c), mutation of the N-box at position −901 resulted in decreased COL1A1 promoter activity. However, disruption of the N-box located at −1740 and −1091 had no influence on activity of the promoter (data not shown). Similarly, mutation of the N-box at position +113 resulted in decreased COL1A2 promoter activity (Figure 4(d)). These results suggest that Hes proteins might augment mouse COL1A1 and COL1A2 promoter activity.
Because Hes1 plays a critical role in regulating lung development, we further investigated whether Hes1 could bind to the mouse COL1A1 and COL1A2 promoters in L929 cells by the ChIP assay using anti-Hes1 (Figure 4(i)). The co-precipitated chromatin DNA fragments were amplified by PCR using primers for mouse COL1A1 and COL1A2 promoters. The results showed that the fragment with the Hes1-binding site was co-precipitated by the anti-Hes1 antibody (Figure 4(i)). This further indicates that the mouse COL1A1 or COL1A2 promoters might be regulated by Notch signaling through Hes1-dependent mechanisms.
We also amplified the human COL1A1 (−2074/+126) and COL1A2 (−1772/+228) promoters and constructed reporter plasmids pGL3-hCOL1A1 and pGL3-hCOL1A2. Gene reporter assays revealed that co-transfection of 293 T cells with NICD gradually increased human COL1A1 and COL1A2 promoter activity (Figure 4(e) and (f)). Mutation of the N-box at position −545 decreased activity of the human COL1A1 promoter (Figure 4(g)). However, disruption of the N-box located at −1890 and −1865 had no influence on the activity of the promoter (data not shown). Mutation of the N-box at position +441 resulted in decreased COL1A2 promoter activity (Figure 4(h)). Similarly, ChIP assays showed that the human COL1A1 and COL1A2 fragments with the previously described Hes1-binding site could be co-precipitated by the anti-Hes1 antibody (Figure 4(j)). Thus, the human COL1A1 and COL1A2 promoters might also be regulated by Notch signaling through Hes1-dependent mechanisms.
Discussion
In the present study, we found that Notch signaling regulates the expression of col1α1 and col1α2 both in mouse L929 cells and in human MRC-5 cells. Aoyagi-Ikeda et al. 19 showed that activation of Notch, either by ectopic expression of the NICD or by the co-culture of RLE-6TN cells (i.e., rat alveolar epithelial cells) with L-Jagged1 cells, induces expression of collagen I in the process of epithelial–mesenchymal transition. Thus, Notch signaling was implicated in the regulation of col1α1 and col1α2 expression in airway fibroblasts.
We identified several Hes binding site-containing N-boxes in the 5′ region of mouse, rat, and human COL1A1 and COL1A2 promoters. This conservation underscored the importance of these motifs as regulatory elements and provided additional novel evidence for the role of Notch signaling in regulating col1α1 and col1α2 expression. Indeed, using luciferase reporter assays, we showed that over-expression of NICD results in activation of the COL1A1 and COL1A2 promoters. Site-directed mutagenesis reporter assays revealed that Hes proteins might increase COL1A1 and COL1A2 promoter activity, and ChIP assays confirmed binding of Hes1 to the COL1A1 and COL1A2 promoter regions. Thus, Notch signaling may regulate col1α1 and col1α2 expression through a Hes1-dependent mechanism.
Several studies have shown that Notch signaling is involved in liver and kidney fibrosis. Chen et al. 13 demonstrated that over-expression of Notch3 in hepatic stellate cells leads to significantly increased expression of a-SMA and collagen I, which suggests that liver fibrosis could be regulated by the Notch pathway. Chen et al. 20 further showed that GSI attenuates hepatic fibrosis via Notch signaling, and other studies showed that Notch signaling plays key roles in kidney fibrosis.14,21,22 Our results expand our understanding of the role of Notch signaling in the fibrosis process.
Hes proteins are usually suppressive basic helix–loop–helix (bHLH) molecules. However, in this study, the binding of Hes proteins increased mouse and human COL1A1 and COL1A2 promoter activity, although the precise mechanism remains unknown. A potential clue comes from two studies showing that Hes1 can switch from a repressor to an activator by direct interaction with cAMP response element-binding (CREB)-binding protein (CBP). 23 Meanwhile, CBP has been implicated in the regulation of COL1A2 promoter activity. 24
The regulation of col1α1 and col1α2 expression by Notch through Hes1-dependent mechanism described here is likely to be relevant in the context of human disease. Failure of this mechanism may be related to airway subepithelial fibrosis, which can lead to fragile asthma. Future experiments will further examine the effects of regulating Notch signaling in airway fibroblasts on asthma in vivo.
In conclusion, this study showed that Notch signaling can directly regulate COL1A1 and COL1A2 promoter activity through a Hes1-dependent mechanism both in mouse L929 cells and in human MRC-5 cells, which could be future targets for pharmacotherapy of airway subepithelial fibrosis in asthma.
Footnotes
Author contributions
All authors participated in the design, interpretation of the studies, and analysis of the data and review of the manuscript. MH and HFOY conceived and designed the experiments, analyzed the data, and wrote the paper, they contributed equally; MH, HFOY, SYQ, and XTX performed the experiments; PW and CGW contributed reagents/materials, and CGW edited the final version of the manuscript.
ACKNOWLEDGEMENTS
The author is particularly grateful to Prof. Hua Han, the Fourth Military Medical University, Xian, China, for his kind provision of the pEF-Bos-NICD plasmid. Dr Feng-Hua Gu and Hai-Ying Wang provided the authors excellent technical assistance. This investigation was supported by National Natural Science Foundation of China (NSFC) (NO. 81170020).