
Abstract
Aging is exemplified by progressive, deleterious changes that increase the probability of death. However, while the effects of age are easy to recognize, identification of the processes involved has proved to be much more difficult. Somewhat surprisingly, research using the budding yeast has had a profound impact on our current understanding of the mechanisms involved in aging. Herein, we examine the biological significance and implications surrounding the observation that genetic pathways involved in the modulation of aging and the determination of lifespan in yeast are highly complicated and conserved.
Introduction
The modern study of aging can be traced back to the coining of the term “gerontology” from the Greek for “the study of old men.” This formalized a field born from the historical discipline of mathematical actuarial science. As early as 1662, it was observed that human longevity often displayed predictable patterns. Subsequently, Edmond Halley, of Halley's Comet fame, proposed that given a large enough sample (Figure 1a), life expectancy and maximum lifespan could be reasonably accurately inferred.
1
Graphical representation of lifespan & mortality rate. (a) Collected longevity data revealing the relative number of people at each specific age in a population at death. (b) The rate at which people within a population die. As age increases, the rate of death increases. (c) Human survival curves reveal that an increase in average life expectancy is observed when data from 1900 is compared to 2006. (Panels a–c adapted from National Vital Statistics Reports, United States Life Tables 2006, vol. 58, #21, 2010). (d). Hypothetical yeast survival curves are shown for both short-lived (SL), wild type (WT), and long-lived (LL) yeast. (A color version of this figure is available in the online journal.)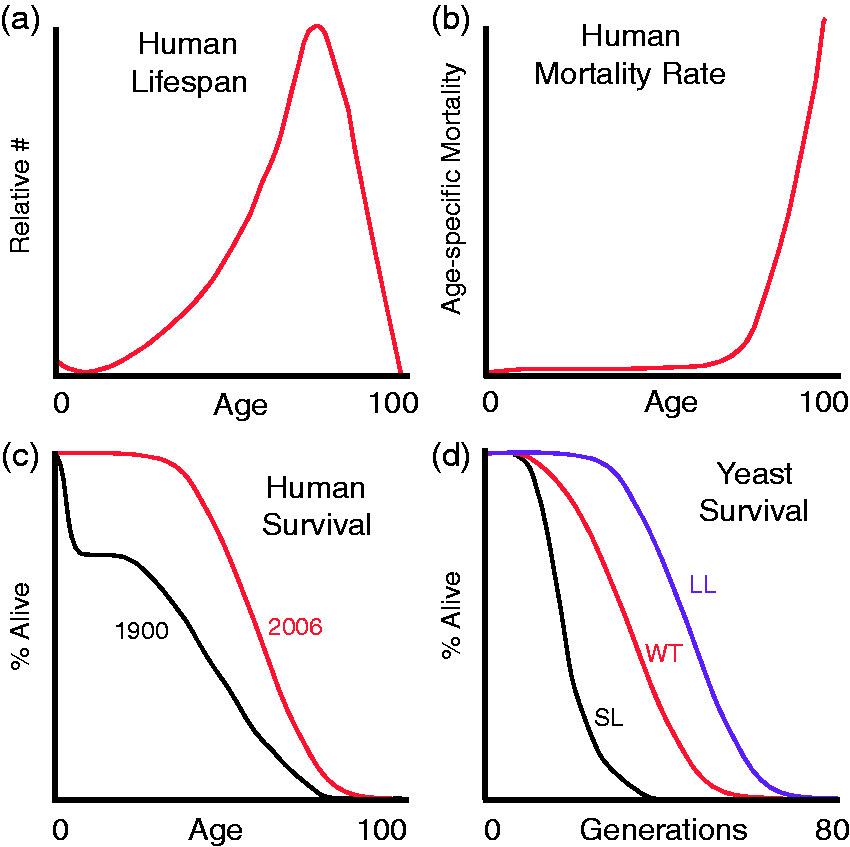
Building off of these observations, Benjamin Gompertz observed that the probability of mortality in adults increases exponentially. Specifically, Gompertz noted that the mortality rate of adults doubles every 7 years (Figure 1b) setting the stage for the development of the first mathematical definition of aging. 2 The Gompertz curve equation, when displayed in its graphical form, first revealed the shape of human longevity (Figure 1c).
Consequently, analyses of systematically collected longevity data revealed that the probability of survival was far from stable or constant. In contrast, human mortality rates are strongly influenced by both genetics and environmental conditions. For example, as medicine and public health care have improved in the past century, the infant mortality rate has also dramatically decreased concurrently extending average life expectancy (Figure 1c). In terms of genetics, a genealogical study of nearly 4000 relatives, Alexander Graham Bell found that the descendants of long-lived parents (e.g. both lived past 80 years of age) lived an average of 20 years longer than descendants of short-lived parents (e.g. both lived less than 60 years of age). 1 Furthermore, somewhat counter-intuitively, it was discovered that an alteration to the environment through a severe reduction in caloric intake (∼40% decrease) substantially prolonged the average and maximal lifespan of laboratory rats. 3 These prescient insights paved the way for what remains today one of the most impactful means of extending lifespan. To date, caloric or dietary restriction is the only non-genetic means that can extend lifespan in almost every organism tested including yeast, flies, worms, and rodents.
Examination of modern longevity data reveal several interesting conundrums. As predicted by the Gompertz-Markham law, the rate of aging of the population can be inferred from the slope of the curves shown in Figure 1(c). Interestingly, with the exception of the infant years, these slopes remain consistently similar. This suggests that the inherent rate of aging of the human population is curiously recalcitrant to the progress of modern medicine and improved health care. Moreover, while medical advances have significantly extended average life expectancies, our maximal observed lifespan remains remarkably unchanged (Figure 1c). Thus, while the mechanisms that modulate human aging are complex, studies in model organisms like the budding yeast have significantly advanced our understanding of the molecular mechanisms involved in longevity.
Studying aging in yeast: a budding field
Mortimer and Johnson developed the yeast model system for aging in 1959 by demonstrating that mortality rates increase with age resulting in cells with a reproducibly finite lifespan. 4 Prior to the 1980s, despite hints from human genealogical studies of genetic determinants for longevity, the generally accepted dogma in gerontology was that aging was too complex to study genetically. Nonetheless, in 1983, a single gene mutation that increased longevity was identified in the nematode C. elegans.5,6 By 1994, the first longevity gene in yeast was discovered. 7
Budding yeast are known to undergo at least three different forms of aging. Replicative lifespan (RLS) is a measurement of aging defined by the number of buds a mother cell produces before death. 4 In contrast, chronological aging involves studying the time non-proliferating cells survive in stationary phase. 8 Furthermore, disruption of the telomerase enzyme complex promotes a third type of aging known as clonal senescence. 9 Herein, we focus on mechanisms involved in the determination of the replicative life span of individual cells.
S. cerevisiae replicate and divide asymmetrically to produce a virgin daughter that is considerably smaller than its mother cell. The original RLS assays relied upon this asymmetry of size and used a microdissection needle to remove daughter cells.
4
Depending upon the strain, mother cells produce ∼20–30 buds before senescing. Throughout most of their life, mothers produce daughters that are noticeably smaller. However, as yeast cells age, they display a number of characteristic morphological and physiological changes as illustrated in the outer circle of Figure 2. These include: an increase in cell size, a wrinkled shape, elongated cell cycle times, fragmented nucleoli, and sterility. Finally, the asymmetry between mother and daughter breaks down. Interestingly, large daughters born to older mothers have a shortened lifespan.
10
Further observations and their implications regarding the regulation of aging in yeast are discussed below.
Factors affecting lifespan in yeast. The outer circle illustrates several morphological and physiological changes in the characteristic of yeast aging, including an increase in cell size and a wrinkled shape at death. The inner circle illustrates six potential or known mechanisms involved in yeast aging. (A color version of this figure is available in the online journal.)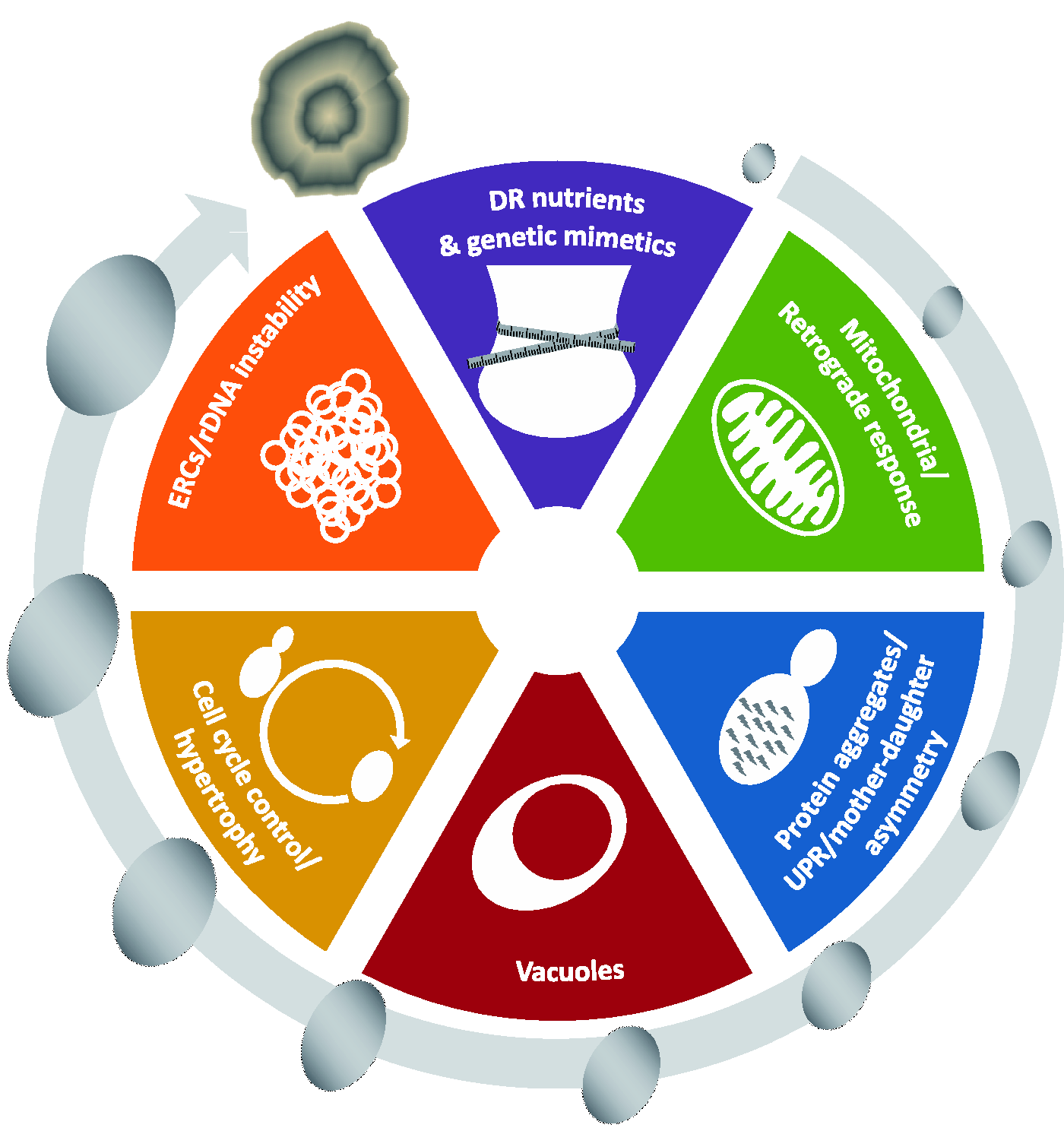
The paradox of rejuvenation and the senescence factor hypothesis
For the most part, in budding yeast, daughter cells are born completely rejuvenated while their corresponding mother cell ages with each bud produced. Unlike higher eukaryotes, budding yeast do not have a separate germ cell lineage. Moreover, the budding proliferation process involves the shared distribution of common cellular components. Specifically, the first building blocks of the virgin daughter cell are inherited directly from the mother. How then does an aged mother produce a fully rejuvenated daughter? The first clues to this paradox have come from observations where daughter cells were born not fully rejuvenated. Early studies revealed that the ability of mothers to generate fully rejuvenated daughters declines with age. 10 Moreover, these “aged” daughters also inherit other morphological phenotypes typically ascribed to older cells: increased size and elongated cell cycle times. 11 Independent studies revealed that in cell:cell fusions the RLS of the zygote is determined by the older cell. 12 In addition, the induction of meiosis and sporulation in older mothers produces fully rejuvenated daughters. 13 Together these studies suggested the existence of diffusible senescence factors.10,14 This theory laid the groundwork for several testable hypotheses. 14 First, senescence factors must accumulate with age. Second, for the most part, senescence factors must be sequestered by mother cells. Third, senescence factors must modulate lifespan such that conditions that increase these factors decrease lifespan and vice versa. Subsequently, researchers set out to test these hypotheses to determine if the accumulation of senescence factors is a driving force in yeast aging. In fact, in this pursuit, most evidence advocates the theory that the retention of senescence factors by mother cells limits lifespan while daughter cell exclusion is the key to rejuvenation. The next key step has been to identify these factors.
ERCs, Sir2, and rDNA instability: early keys to longevity
Extrachromosomal rDNA circles (ERCs) were the first proposed senescence factor. 15 ERCs arise from the 100–200 tandem repeats of ribosomal genes on chromosome XII. Homologous rDNA recombination, promoted by either an open chromatin structure or by stalled replication forks, leads to the production of ERCs. 16 Each ERC contains an autonomous replication sequence, and thus is duplicated in every S phase, leading to an exponential accumulation of circles. Subsequently, it has been proposed that ERCs reduce the proliferative potential of cells by acting as a sink for limiting replication factors. 17 Importantly, the ERC model of aging fits hand-in-hand with the concurrent discovery of SIR2 as an archetypal longevity gene. 18
As the founding member of a family of proteins (sirtuins) conserved from yeast to humans, SIR2 is perhaps the most far-reaching longevity gene identified thus far. In yeast, Sir2 affects the lifespan in direct relation to its expression levels – deletion of SIR2 reduces RLS by ∼50%, while an extra copy of SIR2 extends RLS by 30–40% (Figure 1d). 18 Importantly, deletion of SIR2 increases the amount of ERCs in cells, while overexpression of SIR2 reduces ERC accumulation.18,39 In addition, sporulation eliminates ERCs to rejuvenate cells. 13 ERCs are proposed to be asymmetrically retained by mother cells. However, the mechanism of retention is unclear. One possibility is that ERCs are tethered to nuclear pores. In this model, older nuclear pores with tethered ERCs are sequestered in the mother cell via a septin-dependent diffusion barrier in the nuclear envelope. 20 However, newer studies have shown that daughters freely inherit a mix of older and newer nuclear pores.21,22 Moreover, evidence suggests that the retention of ERCs in the mother is due to the short duration of mitosis and the geometry of the bud neck reducing transference of ERCs to daughters. 23 However, most significantly, exogenous induction of ERCs shortens lifespan. 15 Thus, the accumulation of ERCs represented the first iconic example of an aging factor in yeast.
While it is now evident that Sir2 affects a wide variety of cellular processes, Sir2 was originally identified, and named, by its ability to silence (
Functionally, Sir2 is an NAD+-dependent deacetylase, and thus its activity is linked to intracellular NAD+ concentrations and fluctuations.31,32 The deacetylation reaction produces nicotinamide (NAM), a potent inhibitor of Sir2 activity.31,33,34 In order to maintain Sir2 silencing, cells utilize several different pathways to maintain NAD+ concentration and limit NAM buildup.35,36 In addition, the mechanism whereby Sir2 regulates chromatin depends on the location and involves complexes with many other proteins. For example, Fob1 is involved with the formation of the replication blocks that induce rDNA recombination. 37 As a result, fob1Δ mutants preserve rDNA stability to extend lifespan.38,39 Interestingly, however, it has recently been demonstrated that rDNA instability is sufficient to decrease lifespan even in the absence of ERC accumulation, suggesting that ERCs are more correlative than causative in yeast aging. Furthermore, the central role of Sir2 in yeast was exemplified by the finding that SIR2 deletions suppressed the ability of 32 independent longevity mutants to extend lifespan. This conclusion was reiterated by several QTL genetic mapping studies suggesting SIR2 and rDNA loci account for the majority of lifespan variation in genetically diverse yeast strains.40,41
Following the initial studies in yeast, overexpression of Sir2 orthologs was shown to extend the lifespan in both flies and worms.42,43 However, the functional role of sirtuins in aging is not without controversy. Attempts to reproduce initial observations have yielded contradictory results,44–46 and the specific role of Sir2 in yeast aging has also been debated. 47 In contrast, support for a role for sirtuins in healthy mammalian aging is growing. While increased SIRT1 (the mammalian Sir2 ortholog) expression has not been shown to extend lifespan, SIRT6 does extend the lifespan of male mice. 48 Nevertheless, based upon in-depth analyses of Sir2 function in yeast, it is highly likely that sirtuins are intimately involved in age-associated processes conserved from yeast to man.
Segregating fitness: mother cells giving the best to their buds?
In addition to the asymmetrical segregation of ERCs, new studies also link the asymmetric distribution of old and less-fit organelles or protein aggregates to aging. In particular, observations have revealed that reactive oxygen species (ROS) and oxidatively damaged mitochondria are predominantly sequestered in mother cells.49–53 Specifically, higher levels of ROS are found in dying yeast cells, while new daughter cells tend to have lower levels of ROS even if their mothers had high levels.53,54 Sir2 has a role in the asymmetrical distribution of ROS,50,55 and ROS-induced oxidative damage negatively impacts the lifespan.54,56 Furthermore, cells lacking ROS scavengers have significantly higher levels of DNA damage and display a shortened lifespan. 57
However, the simple conclusion that ROS-induced oxidative damage was directly responsible for aging was considerably weakened by observations that the addition of antioxidants or overexpression of radical metabolizing enzymes did not consistently lead to increased longevity.57–61 In addition, deletion of sod-2 in C. elegans actually increases lifespan despite an apparent increase in oxidative damage. 62 Similarly, the direct induction of ROS also extends lifespan. 63 Strikingly, when SIR2 is deleted, daughter cells have lower levels of ROS compared to WT daughters despite having more fragmented mitochondria and being shorter lived. 53 Additionally, in some cases, ROS levels in wild type yeast increase to high levels even in young cells. 53 Consequently, it has been proposed that ROS and mitochondrial dysfunctions, which increase with age, lead to the activation of a stress response mechanism called mitochondrial hormesis that promotes longevity.64,65
Supplementing this complex relationship is the idea that, although improper segregation of mitochondria shortens lifespan,66,67 yeast strains lacking mitochondrial DNA have an increased lifespan. 68 Evidence suggests that the modulation of lifespan is dependent upon a mechanism of communication between mitochondria and the nucleus called the retrograde response. 69 The retrograde response is a complex signal transduction pathway principally involving Rtg1-3, the transcriptional co-activator SLIK, and the negative regulator Mks1.70–73 While the activity of the retrograde response increases with age,74,75 deletion of RTG2 blocks lifespan extension. 74 Curiously, activation of the retrograde response is associated with increased ERC production. 74 Inactivation of the retrograde response via deletion of GCN5 reduces ERC accumulation, but shortens the lifespan. 76 Thus, evidence suggests that retrograde signaling promotes longevity independent of ERCs.
The mitochondrial unfolded protein response (mtUPR or UPRmt) is another stress-induced mitochondrial-to-nuclear signal transduction pathway.77–79 Initial studies suggest that mtUPR is required for lifespan extension by several longevity mutants in C. elegans.80,81 PHB1 and PHB2 encode the two sub-units of the yeast prohibitin complex involved in mitochondrial protein folding. 82 Deletion of these genes induces mtUPR, but actually shortens lifespan. 83 Thus, the role of the mitochondrial stress response in aging is also clearly complex. The recent discovery of humanin, a potential mitochondria-derived cytoprotective peptide, reinforces the significance of newly discovered mitochondrial stress response pathways. 84 The developing dogma is that mitochondrial signal transduction via numerous outlets has integral roles in aging and fitness.
Asymmetric inheritance of the vacuole is another recently proposed senescence factor. 85 Studies have revealed that mother cells retain vacuoles with a higher pH while daughters inherit vacuoles with lower pH. 52 Vacuoles are involved with the autophagy pathway, in addition to storage of amino acids and ions, and the inheritance of acidic vacuoles is thought to be cytoprotective and extend RLS.85,86 As a mother cells ages, vacuoles increase in size and pH. 87 Disruption of vacuole function and pH reduces the lifespan, while the overexpression of vacuolar proteins such as Osh6 or V-ATPase increases the lifespan. 88 Overexpression of V-ATPase prevents an early rise in vacuole pH, which normally occurs after four or five divisions. 85 It has been suggested that this rise in vacuolar pH releases stored neutral amino acids, decreases mitochondrial function, and promotes aging. 17
Finally, the asymmetric accumulation of damaged protein complexes may also function as senescence factors. 49 Support for this hypothesis comes from the observations that protein aggregates segregate away from the daughter during budding and accumulate in mother cells. 49 This segregation requires Sir2 and the protein disaggregate Hsp104, and it has been linked to actin-dependent processes and the polarisome, which is a complex associated with sites of polarized cell growth.89,90 However, some studies have shown that deletion of the polarisome does not eliminate aggregate segregation. 91 Additional studies have revealed that misfolded, soluble and ubiquitinated proteins accumulate in a spatial compartment called the juxtanuclear quality control (JUNQ) inclusion body 92 while complex protein aggregates accumulate in in a spatial compartment called the insoluble protein deposit (IPOD) inclusion body. 93 JUNQ is associated with the nucleus, and IPOD is associated with the vacuole.92–94 These two inclusion bodies are retained by mother cells during budding while the daughter cell inherits inclusion body-free organelles.92–94 The autophagy pathway has also been linked to segregation of protein aggregates. During meiosis, inhibition of autophagy inhibition prevents sporulation and results in the accumulation of protein aggregates. 91
Dietary restriction and pathways of rejuvenation
In yeast and many other organisms, a reduction in calories to a nutritionally safe state called dietary restriction (DR) results in longevity and increased health span.95,96 Functionally, DR is achieved by reducing the amount of glucose available to cells by 4–20 fold. DR promotes an increase in both the average and maximal lifespan attained by cells suggesting a true change in the rate of aging. Although lifespan extension by DR was first observed in 1935, 3 the mechanisms involved are still not well understood. While DR promotes longevity in nearly all simple model organisms tested, the impact of DR on primate aging is considerably less clear.97,98 Nonetheless, new evidence also suggests that short bursts of fasting, which are more realistically attainable by higher organisms than extended dietary restriction, may be equally capable of activating this DR response.99–101
Original studies in yeast suggested that DR functions through the activation of Sir2. 102 One possible explanation was a DR-induced reduction in the levels of NADH, a repressor of Sir2 activity. 103 NAM, a product of the Sir2 deacetylase reaction, also inhibits Sir2.34,104 The Pnc1 enzyme indirectly activates Sir2 by converting NAM to nicotinic acid. 105 Therefore, a DR-mediated increase in Pnc1 could activate Sir2. 105 However, the hypothesis of a Sir2-mediated DR mechanism has become controversial.106–110 For example, the studies by Lin et al. were conducted in a strain in which the overexpression of Sir2 does not lead to increased longevity.107,109 In contrast, in other strains, overexpression of Sir2 and DR has additive effects on longevity suggestive of parallel pathways. 107 Additionally, in some yeast backgrounds, DR induces longevity in the absence of Sir2, suggesting that Sir2 is not essential for DR-mediated longevity.107,110 Finally, new studies suggest that the reduction in rDNA recombination occurs via a Sir2-independent mechanism.111,112 Thus, the relationship between DR-induced longevity and Sir2 remains unresolved.
Decreasing glucose availability shifts yeast metabolism from fermentation to respiration and extends the lifespan. Concomitantly, cells increase expression of respiration-specific genes, in part due to the HAP transcriptional complex.102,113 Furthermore, autophagy rates increase and growth is slowed. 114 Deletion of HXK2 or overexpression of HAP4 mimics DR to extend lifespan.102,115 In addition, DR reduces the activity of a number of nutrient-responsive signal transduction pathways.
The relationship between the TOR pathway and DR is an area of active research. In the presence of abundant nutrients, TOR is highly active, but when nutrients are scarce, TOR activity is decreased. 116 Inhibition of TOR results in reduced translation initiation and ribosomal biogenesis, but an increase in longevity and stress resistance.117,118 Importantly, repression of TOR activity extends the lifespan in flies, worms, and mice.119–122
Downregulation of the RAS-cAMP-PKA pathway also mimics DR to extend the lifespan. This pathway is normally stimulated by high glucose levels, and loss of function mutations in the G-protein coupled receptor (e.g. gpr1Δ or gpa2Δ) simulate DR to extend lifespan.115,123,124 Furthermore, disruption of PKA activity (e.g. via tpk1, tpk2, tpk3, or cdc25 mutations) or downstream signaling kinases (e.g. sch9 mutation) also induces longevity.115,116 However, the mechanisms responsible for lifespan extension are not well understood.
Recent evidence also implicates vacuole acidity in DR-induced longevity. 85 In young cells DR leads to an increase in vacuolar acidity, while in old cells DR leads to the prevention of a drop in acidity. 85 Support for a vacuolar role in DR is also provided by the fact that when V-ATPase is not functional, DR longevity effects are masked. 85 Furthermore, longevity is not further extended when VMA1, an activator of V-ATPases, is overexpressed. 85
Finally, modulation of ROS production may have a role in DR-induced longevity. In some studies, there is evidence that DR decreases ROS and oxidative damage.52,125,126 However, other evidence suggests that respiration and ROS levels may actually increase under DR conditions. Increased ROS may lead to the activation of a stress response which in turn promotes longevity.52,126,127 Nonetheless, genetic studies have revealed that the mechanisms involved in DR-induced longevity are remarkably complex.
A recent large genetic study exemplified this complexity. 83 Examination of the impact of DR on 166 gene deletions revealed that just less than half (82/166) of these strains responded to DR. 83 Of these 82 deletions, approximately 50% had extended and 50% had reduced lifespans. 83 Lifespan changes ranged from a 79% decrease to a 103% increase. 83 For the most part, DR was very effective at increasing the lifespan of short-lived mutants. 83 However, counter-intuitively, DR was nearly as effective at reducing the lifespan of tested long-lived mutants. 83 Finally, in very few cases did DR extend the lifespan of long-lived mutants. Similar results have been observed in studies in mice and monkeys.128,129 Thus, although there is considerable evidence supportive of DR's ability to promote longevity, DR clearly does not extend the lifespan in every condition tested.
Cell cycle control at the crossroads of reproduction and death
In addition to significantly extending lifespan, DR dramatically extends cell cycle time and reduces cell size (Schneider, unpublished data).83 Furthermore, an examination of 49 long-lived mutants revealed that the majority of these strains had significantly longer cell cycle times. 130 Other studies have indicated a strong correlation between cell size and cycle time.131,132 In general, conditions that elongate cell cycle times tend to produce small cells. These correlations suggest the possibility of a link between cell cycle time/cell size and lifespan. 132 Initial aging studies in yeast reported that a dramatic increase in cell size (hypertrophy) accompanies aging in yeast.132, 133 Nevertheless, a potential causative role for cell size in aging was ruled out when an early study demonstrated that cell enlargement with alpha pheromone did not result in a shortened lifespan. 10 However, attempts to reproduce this experiment have produced contradictory results. 134 In addition, other studies have demonstrated that experimentally increasing the cell size reduces lifespan. 132 Moreover, a correlation has been observed between genetic mutations that modulate cell size and lifespan. 132 In general, mutations that reduce cell size concomitantly increase lifespan and vice versa. 132 Furthermore, mutations that increase the rate of hypertrophy also shorten lifespan. 132 Thus, although cell cycle control, cell size and hypertrophy are regaining entrance into the longevity discussion, there are still many unanswered questions. First, not all long-lived cells have an increased cell cycle time. For example, overexpression of SIR2 extends lifespan but not cell cycle time. 130 Second, conditions that extend cell cycle time can reduce lifespan. For example, in many cases DR shortens lifespan. 83 Third, not all small cells are long-lived or vice versa.135,136 Finally, the biggest missing tenant of a cell cycle/cell size/hypertrophy theory for aging is the lack of mechanistic causation. 137 A number of correlations have been observed, but it is not yet clear whether increasing cell cycle time or decreasing growth rate has a direct role in the modulation of aging.
Reflections and introspections
Now that it has been more than 20 years since the first longevity mutant was identified in yeast,
7
it is a good time to take stock of what has been learned. Many of the genes and pathways discovered in the past 20 years were highlighted in the previous sections. To date, according to the GenAge database,
138
825 genes have been identified that are implicated in yeast aging. These observations suggest that nearly 13% of yeast genes have a role in the regulation of lifespan. Do these numbers reflect the assumed complexity regarding the processes involved in aging, or is it somewhat unexpected to discover that so many single gene deletions strongly alter life expectancy? In most cases where traits are encoded by multiple genes, the depth of genetic redundancy buffers the severity of the observed phenotype. In other words, even without knowing how many genetic pathways are involved in yeast aging, it is not trivial to explain the large number of yeast mutants that have strong aging phenotypes. More specifically, would we have expected the presence of 226 anti-longevity genes in yeast? Thus, in these cases the wild type copy of these genes actually promotes aging, and only loss of function of these genes extends the lifespan. In contrast, at least 20-fold fewer pro-longevity genes have been identified. Furthermore, reevaluation of the shape of the lifespan curves of wild type and mutant yeast strains should reveal a wealth of information about some of the mechanisms involved in yeast aging (Figures 1(d) and 3).
The significance of shape. (a) Human mortality rates plotted on a log scale with values indicated on the right Y-axis. The dotted line represents extrapolated mortality data predicted by Gompertz-Markham law where the mortality rate doubles every ∼8 years. The solid line represents human data points adapted from Ham and Veomett (1980).139 The thicker horizontal line is indicative of an age-independent constant rate of mortality observed in non-aging populations. (b) Various potential lifespan curves that may be generated by the budding yeast as described in the text. (A color version of this figure is available in the online journal.)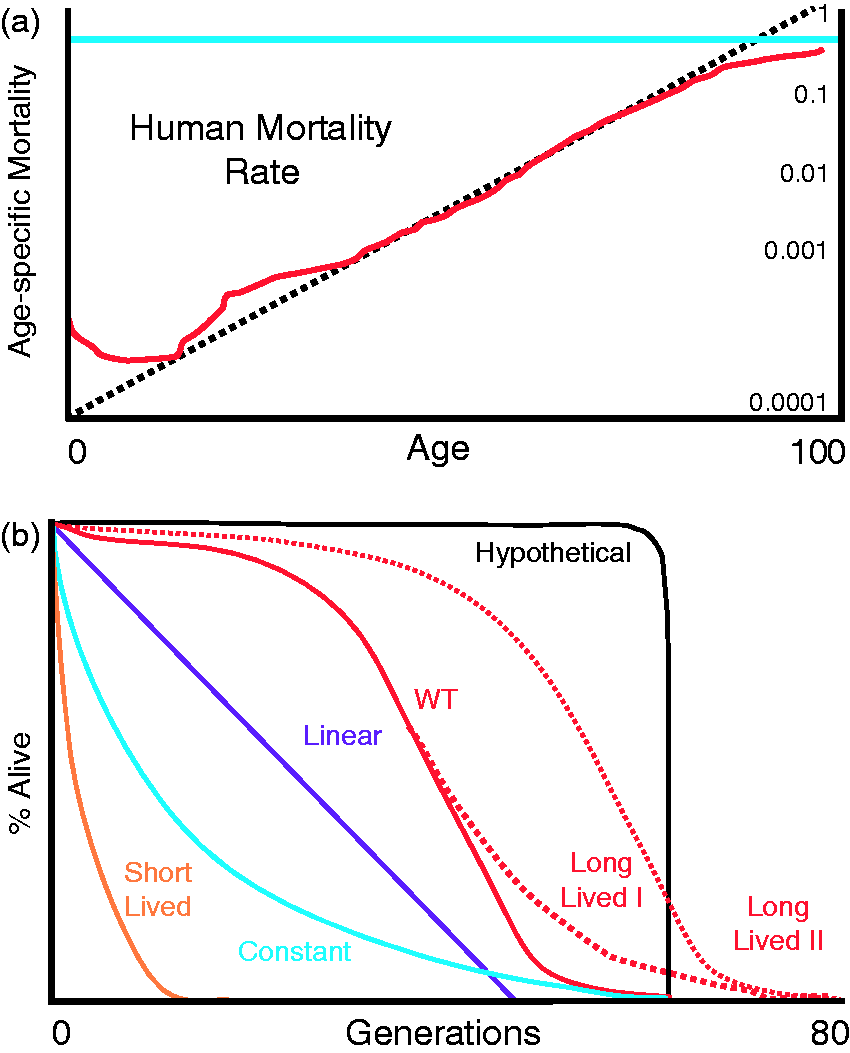
The shape I am in
Plotting human mortality data (Figure 1b) on log scale reveals the nearly straight line predicted by the Gompertz-Markham law (Figure 3a). Two of the three deviations from this trend can be easily explained. A peak in mortality rate in infants is likely due to the inherent fragility of newborns (Figure 3a). A subsequent peak in mortality for adolescents is probably due to high-risk behavior associated with adolescents resulting in more accidental deaths (Figure 3a). The final deviation involves a significant decrease in the mortality rates of the very elderly. This observation is more difficult to explain but could result from the aggregate survival of the fittest individuals.
What insight can be gained by a re-examination of experimental yeast lifespan curves? The characteristic sigmoidal shape of human mortality curves infers a slow–rapid–slow rate of aging (Figure 1c). The vast diversity of the human population in terms of genetic, environmental, economic, and behavioral factors is often proposed to explain the stochastic nature of these curves. Curiously, a simple organism like the budding yeast displays a mortality curve that is nearly identical in shape (Figures 1(d) and 3(b)). Why should genetically identical yeast (WT line in Figure 3b) in a constant and invariant environment also display stochastic mortality? Moreover, the hypothetical expectation for isogenic yeast (Figure 3(b)) has not yet been observed experimentally. The reasons for these observations are not well understood. Nevertheless, there must be a large amount of randomness with respect to the aging process in order to produce this lifespan curve. In other words, some cells must be highly sensitized to aging while others are exceedingly resistant. In theory, this could explain the general shape of aging curves that are observed. By this logic, except for the very few cells that die early, most young cells are not aging, or as affected by aging, as indicated by the initially flat mortality curves. However, as time progresses, a steadily increasing mortality rate is indicative of a response to aging or the onset of an increased rate of aging. Late in life, for yet unknown reasons, either the rate of aging decreases or cells become less susceptible to the effects of age.
Using this framework, what can be made of mutations and conditions that alter lifespan? Examination of mortality rates reveals that the slope of the linear part of lifespan curves reflects aging rates; the steeper the slope, the sooner the mortality rates peak. In the ideal case of an exponentially increasing mortality rate, the ensuing lifespan curve would be predicted to be linear (Figure 3b). Similar lifespan curves have been seen in rad6Δ and rpt1sΔ mutants.30, 140 Nonetheless, these cells appear to be aging slower than wild type cells. Conversely, in the complete absence of aging, lifespan curves would display the consistency of a decaying exponential (constant line in Figure 3b). This pattern is observed in the half-life of radioactivity, and has been observed in several yeast mutants.139,140 Importantly, some conditions or mutations (whi5Δ or fob1Δ) could slow aging entirely (e.g. observe the decreased slope of the Long Lived I line). In contrast, parallel slopes indicate a time response. Thus, some conditions or mutations could delay (Long Lived II line as observed in rpl42aΔ or ssf1Δ 132 ) or advance (short lived line as observed in sir2Δ and cln3Δ132,142) the window of susceptibility to aging. In cases of extremely rapid aging, one would expect to see very high mortality rates at all ages resulting in extremely short lifespans.
Summary
In yeast and all organisms, it is essential that the inherited lifespan of offspring is largely independent of parental age. Otherwise, the transfer of accumulated damage to descendants, via some sort of Lamarkian model, would be highly detrimental. In yeast, lifespan is nearly always rejuvenated in daughters despite increasing age in mothers. This observation is really quite amazing given that mothers and daughters share cellular contents. Years of research have begun to elucidate how lifespan is rejuvenated each generation. Nevertheless, it is still not well understood what causes this mechanism to break down in older yeast or in the few daughters that die young. Thus, lifespan regulation is clearly robust but at the same time highly variable. Small events (e.g. drifting environmental conditions or single gene mutations) can have profound effects, supporting the conclusion that aging is regulated by a myriad of intersecting genetic pathways.
Since the paper was written and submitted for publication, new and exciting work has been published showing the effect of ibuprofen and aromatic amino acid uptake on lifespan extension and cell size. 143
Footnotes
Authors' contributions
All authors participated in the writing and review of the manuscript.
Acknowledgments
The authors thank Nadia Tishler for editing and proofreading. This manuscript was not funded by a grant from any funding agency in the public, commercial, or not-for-profit sectors. JTS, JW and BLS were supported by funds from TTUHSC.