
Abstract
It has been shown in many clinical studies that the level of vascular endothelial growth factor-C (VEGF-C) positively correlates with lymph node metastasis. Nevertheless, beyond the canonical role of VEGF-C in stimulating lymphangiogenesis and thus promoting lymph node/distant metastasis, emerging evidence indicates that expression of VEGF-C contributes to various aspects of carcinogenicity via autocrine regulation. The newly identified functions of VEGF-C include but are not limited to proliferation, migration, invasion, and chemo-resistance. Besides tumor cell autocrine regulation, VEGF-C can also modulate the immune system such that tumor cells more easily escape immune surveillance. Therefore, understanding the functional roles and regulatory mechanisms related to the VEGF-C axis may lead to alternative strategies for cancer treatment. This mini-review will focus on summarizing recent discoveries regarding the unconventional functions of VEGF-C in cancer progression.
Keywords
Introduction
Vascular endothelial growth factor-C (VEGF-C) belongs to the VEGF family which, in mammals, consists of VEGF-A, placental growth factor, VEGF-B, VEGF-C, and VEGF-D. 1 Each member of the VEGF family is encoded by a different gene but shares the VEGF homology domain. 2 Among the VEGF family members, VEGF-C shares the highest homology with VEGF-A (∼30%), 3 which is considered to be a critical angiogenic factor. VEGF-C, on the other hand, acts mainly in development- and disease-associated lymphangiogenesis. This mini-review will briefly introduce the conventional role of VEGF-C and summarize recent findings of the unconventional functions of VEGF-C and its receptors in cancer progression.
The conventional role of VEGF-C
The gene for VEGF-C was originally cloned from a cDNA library derived from the human prostatic adenocarcinoma cell line PC-33 and encodes a protein that requires proteolytic cleavage to produce a functional secreted protein. In embryonic development, VEGF-C is required for the initial migration and sprouting of the committed endothelial cells from the primary lymph sac. Knockout of Vegfc causes prenatal death of mice due to a lack of lymphatic vessel formation. 4 During development of the lymphatic system, expression of the tyrosine kinase receptor VEGFR-3 (FLT4) is gradually confined to lymphatic endothelial cells. This receptor is activated by the binding of VEGF-C that is produced by adjacent regions, triggering downstream signaling that can promote proliferation, migration, and survival of lymphatic endothelial cells.5,6 In addition to VEGFR-3, fully processed forms of VEGF-C can also bind to VEGFR-2, which is also expressed on endothelial cells and activation of VEGFR-2 is mainly involved in angiogenesis. 3 Besides binding to the well-known VEGFR, VEGF-C also binds to another group of transmembrane proteins, the neuropilin (NRP) family. NRP1 and NRP2 are co-receptors bound by the class 3 semaphorin (SEMA) and VEGF families, which modulate plexin and VEGFR signaling in vascular and neural development. 7 VEGF-C binds preferentially to NRP2 compared to NRP1, and the binding of VEGF-C to NRP2 enhances VEGFR-2 or VEGFR-3 activity. 8 NRP2 is required for the formation of small lymphatic vessels and capillaries 9 and the expression of NRP2 augments the effects of VEGF-C-VEGFR-3 axis in lymphangiogenesis. 10 The binding of VEGF-C to receptors that are expressed on lymphatic and blood vessels results in both angiogenesis and lymphangiogenesis, reflecting the complexity of VEGF-C signaling.
Pro-lymphangiogenic function of VEGF-C in cancer
Increased expression of VEGF-C in tumors and higher levels of VEGF-C in serum of cancer patients have been observed.11–13 Enhanced expression of VEGF-C is prevalent in lymph node positive tumors and is associated with high lymphatic vessel density and poor survival such as in breast, prostate, and colon cancer.14–19 The originally identified function of VEGF-C in cancer is similar to what has been observed in embryonic development as overexpression of VEGF-C in tumor cells induced peritumoral and intratumoral lymphangiogenesis in mouse models of breast cancer. 20 , 21 It is evident that increased tumor-associated lymphangiogenesis is sufficient to promote metastasis to regional lymph nodes and to distant organs.20–22 Interestingly, in addition to promoting tumor-associated lymphangiogenesis and angiogenesis, growing evidence suggests that VEGF-C can also contribute to tumor progression by means of autocrine signaling.
Autonomous function of VEGF-C and receptors axis in cancer
Solid tumors metastasize frequently through the lymphatic system and regional lymph nodes. Overexpression of VEGF-C in cancers suggests a role of the protein in promoting lymph node metastasis via increased lymphangiogenesis. While the expression of VEGF-C receptors is limited to lymphatic endothelial cells (LECs) during development, in the context of cancer, re-expression of those receptors on tumor cells further suggests the existence of autocrine regulation by VEGF-C.
VEGF-C and VEGFR
Increased VEGF-C and VEGFR-3 expression was first demonstrated in acute myeloid leukemia (AML), the hematological cancer, suggesting a possible function of VEGF-C beyond stimulation of lymphangiogenesis. 23 Subsequent studies showed that VEGF-C can signal through VEGFR-3 expressed on the leukemic cells to regulate the pro-survival protein Bcl-2 and affect both proliferation and response of tumor cells to chemotherapy. 24 A very recent study in ovarian cancer also showed that inhibition of VEGFR-3 sensitized tumor cells to chemotherapy, an action which involves down-regulation of the tumor suppressor gene BRCA1/2. 25 In addition, VEGF-C and/or VEGFR-2/3 signals have been linked to proliferation, migration, and invasion of gallbladder cancer, esophageal cancer, breast cancer, and lung adenocarcinoma cells.26–29
VEGF-C and neurophilin
The VEGF-C/NRP2 axis has recently been implicated in enhancing cell survival under oxidative stress and therapeutic-induced autophagy in prostate cancer cells, while different mTOR is involved as downstream effector.30,31 In a breast cancer model, a similar antioxidant function of VEGF-C/NRP2 was observed. However, in this study, superoxide dismutase 3 was identified as the principal mediator. 32 It should be noted that NRP2 lacks an intrinsic catalytic domain in the cytoplasmic tail, and therefore, no clear evidence suggests that NRP2 is capable of transducing downstream signal alone. Instead, NRP2 is required to cooperate with either conventional (plexins and VEGFRs) or unconventional receptors (such as integrins). 33 NRP2 has been shown to mediate TGFβ-induced epithelial-to-mesenchymal transition (EMT) in lung cancer cells and is required for breast tumor initiation via stimulation of integrin and focal adhesion kinase.34,35 However, the potential involvement of VEGF-C in NRP2-mediated phenotypes in these studies was not discussed.
Emerging role of VEGF-C in cancer stemness
Selective tumor cell-autonomous function of VEGF-C axis in human cancer

NRP: neuropilin; VEGF-C: vascular endothelial growth factor-C.
Immune regulatory role of VEGF-C in tumor microenvironment
The tumor microenvironment is generally considered to be associated with inflammation, and infiltrating immune cells are thought to provide a source of cytokines and growth factors which can promote tumor growth. Besides the known functions in regulating endothelial and cancer cells, VEGF-C was recently found to be expressed by immune cells or to regulate immune response. Expression of VEGF-C can be induced in tumor-associated macrophages by inflammatory cytokines secreted from tumor cells, and increased VEGF-C can in turn promote tumor-associated lymphangiogenesis and metastasis.38,39 A negative correlation of VEGF-C with tumor-infiltrating dendritic cells has been observed in gastric cancer, 40 implying a role for VEGF-C in immune modulation. Natural killer (NK) cells from AML patients express higher levels of VEGFR-3 and inhibition of VEGFR-3 restored cytotoxic activity of the AML-NK cells41,42 further supporting the importance of VEGF-C-VEGFR-3 axis in tumor immunity. Another study in a mouse mammary carcinoma model showed that knockdown of VEGF-C switches the infiltration and phenotype of T cell and dendritic cells toward an antitumor environment. 43 Similarly, VEGF-C has been demonstrated to promote immune tolerance via suppression of CD8+ T cells in draining lymph nodes of melanoma. 44 Together, these studies reveal a potential function of VEGF-C as an immunomodulator and provides a mechanism by which tumor cells escape immune surveillance.
Regulation of VEGF-C expression
In mouse embryos, VEGF-C mRNA expression was enriched in mesenchymal cells and regions where lymphatic vessels were sprouting. 45 Although the mechanism by which VEGF-C is expressed in development is largely unknown, studies in cancer provide evidence for how VEGF-C can be regulated.
Transcription factor
Homeoproteins are transcription factors that are highly conserved among species and are considered to be the master regulators in embryogenesis. The expression of prostate-specific homeobox protein NKX3.1 is lost in prostate cancer and has been shown to inhibit VEGF-C expression by cooperating with histone deacetylase. 46 Sine oculis homeobox homolog 1 (SIX1), which is highly expressed in metastatic breast cancer cells, activates VEGF-C transcription and promotes lymph node metastasis. 47 Many other transcription factors, such as chicken ovalbumin upstream promoter transcription factor 2 (COUP-TFII), inhibitor of differentiation/DNA binding factor (Id-1), Rho GDP dissociation inhibitor 2 (RhoGDI2), and high mobility group box 1 (HMGB1) can also regulate the expression of VEGF-C.48–51 However, whether the regulation is mediated by direct binding to the VEGF-C promoter warrants further investigation.
Cytokines, growth factors, and extracellular matrix (ECM)
Analysis of the VEGF-C promoter led to the identification of putative binding sites for NFκB, implying a mechanism by which inflammatory stimuli may control VEGF-C expression. 52 Indeed, pro-inflammatory factors such as interleukin 1β (IL-1β) and IL-6 increased VEGF-C mRNA expression 53 ; in addition, growth factors such as PDGF, EGF, or TGFβ can also activate VEGF-C expression.54,55 Co-expression of cyclooxygenase (COX)-2 and VEGF-C has been reported in several cancers and further evidence indicates that COX-2 derived prostaglandin E2 (PGE2)-EP receptors is another mechanism by which VEGF-C expression is regulated.56–58
Components in the ECM have also been shown to regulate VEGF-C expression. Heparanase, an endoglycosidase which cleaves heparan sulfate in the ECM, participating in ECM remodeling and degradation, can stimulate VEGF-C expression and promote tumor lymphangiogenesis. 59 Furthermore, induction of VEGF-C by extra domain A, an alternative splicing domain of fibronectin, has been shown in colorectal cancer cells. 60 Overall, VEGF-C expression can be activated by a wide range of signaling pathway including, P38-NFκB, phosphoinositide 3-kinase-Akt, or MAPK signaling, though the specific mechanisms depend on the individual stimuli and cellular context.53,61,62
Post-transcriptional, translational, and epigenetic regulation
MicroRNA is an emerging mechanism by which gene expression is regulated at a post-transcriptional level. Tumor suppressor microRNAs, miR-1862, miR27b, and miR12863–65 have been found to target VEGF-C and inhibit tumor progression. While there is no putative hypoxia responsive binding site on the VEGF-C promoter and a previous study showed that VEGF-C mRNA does not change under hypoxic conditions, 54 a very recent study demonstrated that VEGF-C protein is increased in tumors and this is mediated in an internal ribosome entry site (IRES)-dependent manner. 66 In addition, it has been found that in gastric cancer cell lines, expression of VEGF-C was inversely correlated with VEGF-C promoter methylation status. Moreover, treatment with a methyl donor S-adenosylmethionine induced VEGF-C methylation, leading to a decrease in VEGF-C expression. 67 However, whether hypomethylation of VEGF-C leads to its overexpression in other types of cancer remains to be evaluated.
It should be noted that the specificity and activity VEGF-C can be regulated by post-translational processing (proteolytic cleavage). 68 Proprotein convertase furin, PC5, and PC7 have all been identified as VEGF-C candidate convertases, 69 providing an additional mechanism by which VEGF-C can be functionally regulated.
Conclusion and perspectives
VEGF-C is an important factor in promoting lymphangiogenesis and lymph node metastasis of cancer cells. In addition, VEGF-C displays multifunctional roles in cancer progression and thus warrants increased attention. Because VEGF-C can affect endothelial cells, tumor cells, and immune cells (Figure 1), researchers are seeking methodologies to block VEGF-C signaling by various mechanisms, including an anti-VEGF-C antibody, a soluble receptor mimic that traps VEGF-C, an antibody that interferes with VEGF-C/receptor interaction, or inhibitors that target VEGFR tyrosine kinase activity.70–74 While targeting the VEGF-C-receptor axis is a direct way to inhibit VEGF-C mediated effects, understanding the mechanism by which VEGF-C is up-regulated in cancer may provide additional strategies for cancer treatment. For example, since COX-2-PGE2 regulates VEGF-C expression, inhibitors of COX-2 may also effectively inhibit VEGF-C-mediated lymphangiogenesis and metastasis.
Multiple functions of VEGF-C in tumor progression. VEGF-C binds to receptors expressed on endothelial cells and promotes angiogenesis and lymphangiogenesis. Tumor cells which express VEGFR-2, VEGFR-3, and NRP2 can receive VEGF-C autocrine signals and trigger downstream pathway which mediates aggressive phenotypes. Tumor-associated macrophages are another source of VEGF-C which contributes to the increased expression of VEGF-C in the microenvironment. Immune cells, such as NK cells, can receive VEGF-C signal and thus exhibit immune suppressive functions which favor tumor progression. VEGF-C: vascular endothelial growth factor-C. (A color version of this figure is available in the online journal.)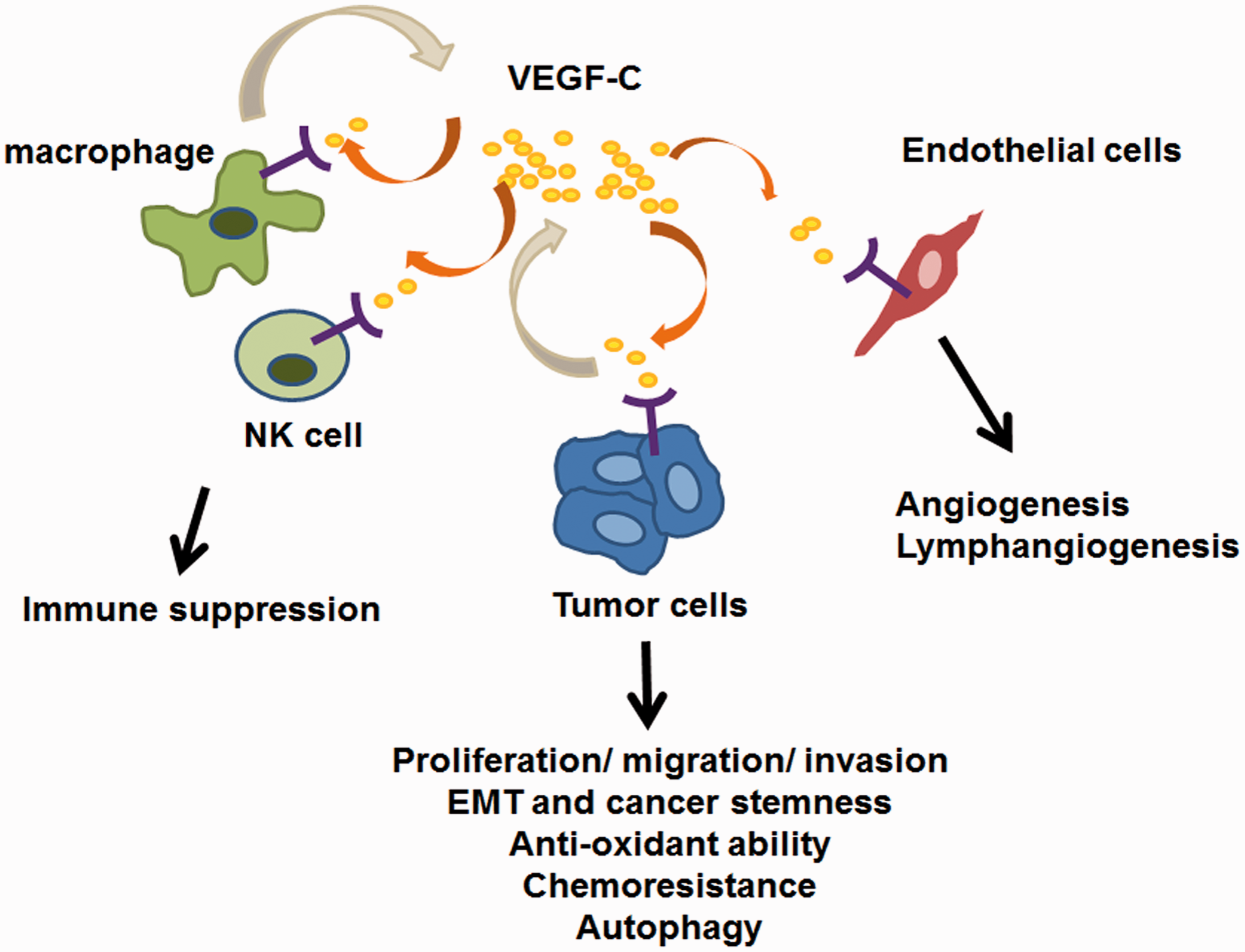
In breast cancer, overexpression of VEGF-C is known to be associated with lymph node metastasis.75,76 Moreover, a recent study showed that VEGF-C mRNA expression is specifically enriched in the claudin-low subtype of breast cancer, an aggressive subtype that confers poor prognosis and for which almost no target therapy exists. 32 Since inhibition of VEGF-C sensitizes tumor cells to chemotherapies, it is possible that inhibition of VEGF-C may increase current treatment efficacy specifically in this subtype of breast cancer. Therefore, combination of anti-VEGF-C with other therapies may be applied to cancer patients even when lymphangiogenesis/lymph node metastasis is not involved.
Current evidence indicates VEGF-C plays an important role in promoting cancer progression via paracrine and autocrine mechanisms, making VEGF-C axis an attractive target to inhibit tumor progression. Additionally, VEGF-C expression has been demonstrated to sensitize lymphatic endothelial cells to radiation-induced cell quiescence, which may account for lymphedema in breast cancer patients after radiotherapy. 77 Therefore, blocking VEGF-C signaling in conjunction with other treatments as anticancer therapy may be able to “kill two (or more) birds with one stone.” However, since VEGF-C binds to multiple receptors to trigger different downstream signaling pathways and up-regulation of VEGF-C has been observed in tumor cells that have acquired resistance to anti-VEGF therapy, 78 targeting VEGF-C as an anticancer therapy may be more difficult than one would anticipate. Therefore, potential disadvantages of anti-VEGF-C therapeutics should be taken into consideration when it comes to clinical application.
Footnotes
Authors’ contributions
CA-W drafted and edited the manuscript. SJ-T edited and finalized the manuscript.
ACKNOWLEDGEMENTS
We apologize to those authors whose work has not been cited. We thank Dr Marcus J. Calkins for proofreading of this current manuscript. Chu-An Wang is funded by grant from Ministry of Sciences and Technology, Taiwan (MOST 103-2321-B-006-020-MY3). Shaw-Jenq Tsai is funded by grants from National Research Program for Biopharmaceuticals (NSC 101-2325-B-006-017), National Health Research Institute (NHRI-EX-102-10244BI), and Top University grant of National Cheng Kung University (D103-35A17).