
Abstract
MicroRNAs (miRNAs) play an important role in regulation of gene silencing and are involved in many cellular processes including inhibition of infected viral replication. This study investigated cellular miRNA expression profiles operating in response to influenza virus in early stage of infection which might be useful for understanding and control of viral infection. A549 cells were infected with different subtypes of influenza virus (pH1N1, H3N2 and H5N1). After 24 h post-infection, miRNAs were extracted and then used for DNA library construction. All DNA libraries with different indexes were pooled together with equal concentration, followed by high-throughput sequencing based on MiSeq platform. The miRNAs were identified and counted from sequencing data by using MiSeq reporter software. The miRNAs expressions were classified into up and downregulated miRNAs compared to those found in non-infected cells. Mostly, each subtype of influenza A virus triggered the upregulated responses in miRNA expression profiles. Hsa-miR-101, hsa-miR-193b, hsa-miR-23b, and hsa-miR-30e* were upregulated when infected with all three subtypes of influenza A virus. Target prediction results showed that virus infection can trigger genes in cellular process, metabolic process, developmental process and biological regulation. This study provided some insights into the cellular miRNA profiling in response to various subtypes of influenza A viruses in circulation and which have caused outbreaks in human population. The regulated miRNAs might be involved in virus–host interaction or host defense mechanism, which should be investigated for effective antiviral therapeutic interventions.
Introduction
The pandemic influenza virus usually infects a large population, not only human but also livestock, such as swine and poultry, resulting in problems in public health and loss of economic distress in terms of hospitalization.1,2 More than 284,000 people worldwide died because of pH1N1 after a year of pandemic. 3 Some strains of influenza virus can be zoonotic, such as H5N1, leading to great losses in the poultry industry, and with high mortality rate in reported human cases (>60%).4–8 Unlike being infected with seasonal influenza, H5N1 infection can cause multi-organ failure, hemorrhagic syndrome and cytokine storm, suggesting that host genes can respond in a different manner depending on the specific strain of influenza virus infection. Previous studies showed that influenza virus principally controls host genes to facilitate viral replication in multiple steps, and one of the potential mechanisms to control gene expression is microRNA (miRNA) regulation.9–13
MiRNAs are small (17–24 nt in length), endogenous, non-coding RNAs found in multicellular organisms. 14 They are acknowledged as vigorous tool regulator molecules that can trigger the targeted gene functions by the translational repression or mRNA degradation through the RNA interference pathway.14–18 Previous studies showed that the genome of more than 150 species including viruses contain sequences of mature miRNAs, especially in the human genome, in which more than 1000 miRNAs have been characterized.16,19–21 Studies have expressed that a broad range of cellular mechanisms are mediated by miRNAs, such as cell proliferation signaling, chromosome maintenance, cell differentiation and apoptosis.9,14,22–24 Not only can viruses regulate host gene expression by using miRNAs to promote the viral life cycle, but the host cell can also alter differential gene expression to respond against viral infection via the miRNA mechanism. The proportion of the miRNA level due to the encounter between host and virus will indicate the disease progression and outcome.25,26 Multiple pieces of evidence have shown that both host and virus constructed miRNAs are essential for virus propagation. It has been reported that the influenza virus plays a critical role in the modulation of the miRNA mechanisms of infected cells and animal models.9,27–29 In contrast, miRNAs can also react with the influenza virus replication process. Many previous studies have mentioned that influenza virus replication and propagation were inhibited due to cellular miR-323, miR-491, miR-654 and miR-146a expression which specifically bind to the PB1 gene of the virus.30,31
Many techniques have been used to validate the miRNA screening and target prediction of miRNA response to influenza virus infection, such as qPCR, luciferase assay and drug compound library screening.9,12,28,31 Among these, microarray techniques have been used extensively to study the miRNA profiles in response to the pathogenesis of the influenza virus.32–35 Although microarray technique can gain a large amount of data of miRNA expression, it cannot determine the significance of particular genes in the cellular mechanism which are not covered by the probe set, and may lead to some essential genes, which slightly change, being dismissed. The high-throughput next-generation sequencing (NGS) can cope with these problems, as the dataset generated from NGS will demonstrate the genome-wide miRNA expression profiles of infected host cells. This study aimed to investigate cellular miRNA expression profiles operating in response to three specific strains of influenza A virus responsible for causing outbreaks in the human population; Avian-origin highly pathogenic avian influenza (HPAI) H5N1, human pandemic (pH1N1) and seasonal H3N2 influenza A virus infection. The study also aimed to predict target genes which might be useful for understanding the host defense mechanism in terms of regulating viral infection.
Materials and methods
Cell culture
A549 (human lung carcinoma) cells used in this study were kindly provided by Prof. Jen-Ren Wang, Department of Medical Laboratory Science and Biotechnology, Center of Infectious Disease and Signaling Research, National Cheng Kung University. A549 cells were cultured in Dulbecco’s modified eagle medium (DMEM) (Thermo Scientific) containing 10% heat inactivated fetal bovine serum (Thermo Scientific) and 1% (v/v) antibiotic/antimycotic (Gibco) under humidified atmosphere with 5% CO2 at 37℃.
Virus infection
A549 cells were seeded (3 × 105 cells/well) in DMEM medium without antibiotic/antimycotic in 6-well plates. Then influenza A viruses subtype pH1N1 (A/Thailand/104/2009), H3N2 (A/Thailand/CU-H187/2010) and H5N1 (A/Thailand/NK165/2005) were prepared by diluting the virus stocks with DMEM medium to yield the desired concentration of 3 × 105 pfu/mL (MOI = 0.5). Each viral infection was performed in triplicate in a biosafety level 3 (BSL3) laboratory, Faculty of Medicine, Chulalongkorn University. When cells reached approximately 80% confluence, cell culture media were removed from each well and cells were washed with 3 mL of phosphate buffer saline (PBS). After that 500 µl of each virus suspension was added into each well and then incubated in a humidified atmosphere with 5% CO2 at 37℃ for 1 h with occasional shaking to allow the virus to adsorb into the cells. After incubation, the virus suspension was removed from each well and then washed by 3 ml of PBS. Finally, 3 ml of complete DMEM medium was added into each well and cells were incubated for 24 h. The protocol of this study was approved by the Institutional Review Board (IRB No. 121/55), Faculty of Medicine, Chulalongkorn University.
Library preparation
After 24 h post infection, triplicate wells were pooled together and microRNAs were extracted by using a microRNA purification kit (Norgen) following the manufacturer’s protocol. The concentrations of microRNA were quantified by using Qubit fluorometer (Invitrogen) with a Quant-iT™ RNA Assay kit (Invitrogen). Purified microRNA (0.2 µg) from each group (uninfected cells, pH1N1 infected cells, H3N2 infected cells and H5N1 infected cells) were used to construct four libraries with different indexes according to a TruSeq Small RNA sample preparation kit (Illumina). The length of DNA libraries were validated by using Agilent 2100 Bioanalyzer system (Agilent) with a high sensitivity DNA chip (Agilent). The concentrations of DNA libraries were quantified by using Qubit fluorometer (Invitrogen) with a Quant-iT™ DNA Assay kit (Invitrogen). The four DNA libraries were pooled together with equal concentration and then high-throughput sequenced using a MiSeq platform (Illumina).
Analysis of microRNA expression profiles
MiSeq reporter software version 2.4 was used for analysis of sequencing data. Low-quality reads (Q-score < 30) were excluded whereas low-quality regions of sequences were trimmed. The passing filtered reads (Q-score ≥ 30) were aligned with human genomic DNA (hg19), mature & precursor human miRNA (from miRbase) and contaminant RNA (human tRNA, rRNA and mRNA) using the Bowtie algorithm (http://bowtiebio.sourceforge.net). The clusters which matched to the miRNA database but not contaminant RNA were considered as miRNA. The miRNAs were classified and counted based on the number of reads matched to the miRbase (www.mirbase.org/).
Differential expression analysis and data visualization
The data of validated miRNAs expressed in cell culture obtained from next-generation sequencing platform were demonstrated in form of fold change, compared to the amount of those miRNAs expressed in control experiment (mock infection). Differential expression analysis was calculated in terms of fold changes
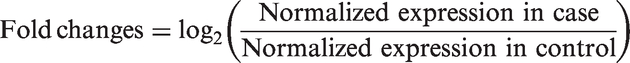


The data visualization was performed by using the GraphPad Prism 5 software to demonstrate the up-regulated and deregulated miRNAs.
Analysis of microRNA-target genes and biological process classification
Identifications of target genes for selected miRNAs were analyzed by using the experimentally validated miRNA-target interactions data accumulated in miRTarBase version 4.5 (http://mirtarbase.mbc.nctu.edu.tw/). Briefly, the target genes of miRNAs were selected based on strong experimental evidence obtained from validation methods including reporter assay, qPCR or western blot. The PANTHER (Protein ANalysis THrough Evolutionary Relationships) classification system version 9.0 (http://www.pantherdb.org/) 36 was used to classify genes and proteins in order to facilitate high-throughput analysis. Each of the validated target genes of miRNA were classified based on biological processes including apoptotic process (GO:0006915), biological adhesion (GO:0022610), biogenesis (GO:0071840), biological regulation (GO:0065007), cellular process (GO:0009987), developmental process (GO:0032502), immune system process (GO:0002378), localization (GO:0051179), multicellular organismal process (GO:0032501), metabolic process (GO:0008152), reproduction (GO:0000003), and response to stimulus (GO:0050896).
Prediction of protein–protein interaction
The target genes of significant miRNAs from NGS experiment were performed for protein interaction. The interactions among the proteins in this study were analyzed from the STRING 9.1 database,37,38 which merges and weighs information from various sources, covering conserved neighborhood, gene fusions, phylogenetic occurrence, co-expression, experiments, database imports, and text-mining. 39 The scores greater than 0.7 are considered as the high confidence.
Results
Analysis of human miRNAs by NGS based on MiSeq platform
After 24 h of infection, triplicate wells of each group were pooled together in order to generate 4 DNA libraries including uninfected control, pH1N1, H3N2 and H5N1 influenza A virus infected cells. Then the 4 libraries with different indexes were subjected to high-throughput sequencing using MiSeq platform. A total of 3,654,615 reads were obtained from a high-throughput sequencing process. After data filtering based on quality score (Q ≥ 30), 2,225,781 high-quality filtered reads were obtained and used for further analysis. The overall reads with a perfect match compared to sequences of miRNA database (miRBase version 16.0) (www.mirbase.org/) were characterized and counted. According to miRBase data, the results showed that there were totally 19,732 reads belonging to human miRNAs, including 4604, 6482, 4509 and 4137 of identified reads which were obtained from non-infected, pH1N1-infected, H3N2-infected and H5N1-infected cells, respectively.
Differential expression of miRNA profiling
MiRNAs expression profiles of A549 cells infected with each subtype of influenza A virus were compared with uninfected cells. The list of differentially expressed miRNAs was identified and classified as upregulated (>1.5 fold increase) and downregulated (<1.5 fold decrease) compared to those miRNA levels in uninfected cells. The full list of miRNA profiling is shown in Figure 1. A549 infected with pH1N1 influenza A virus expressed 10 downregulated miRNAs and 29 upregulated miRNAs. Infection of H3N2 influenza A virus showed seven downregulated miRNAs and 20 upregulated miRNAs while A549 cells infected with HPAI H5N1 influenza virus revealed five downregulated and 23 upregulated miRNAs. The miRNA expression profiles of cells infected with each subtype of influenza A virus were compared both in upregulated (Figure 2a) and downregulated (Figure 2b) miRNAs in order to identify common miRNA responses to multiple subtypes of influenza A virus. The results showed that four miRNAs including hsa-miR-101, hsa-miR-193b, hsa-miR-23b and hsa-miR-30e* were found to have upregulated expression when infected with all three strains of influenza A virus. The hsa-miR-128, hsa-miR-130a, hsa-miR-181c, hsa-miR-301a, hsa-miR-127-3p and hsa-miR-769-5p were upregulated in cells infected with H3N2 and H5N1 while let-7e and hsa-miR-15b were upregulated by pH1N1 and H5N1 infection. For downregulated miRNAs, hsa-miR-125b was downregulated by H5N1 and pH1N1 infection while only hsa-miR-484 was downregulated by H5N1 and H3N2 influenza viruses.
The miRNA expression profiles in each subtypes of influenza A virus infection. A549 cells were infected (MOI = 0.5) with three subtypes of influenza virus. (a) Seventy-eight miRNAs were expressed after infected with pH1N1 influenza virus, including 10 downregulated miRNAs, 29 upregulated miRNAs and 39 miRNAs that expressed in the similar level as control cells. (b) Infection of H3N2 influenza virus showed total of 95 miRNAs expression, including seven downregulated miRNAs, 20 upregulated miRNAs and 68 miRNAs that expressed in the similar level as control cells. (c) Ninety-nine miRNAs were responded in A549 cells infected with HPAI H5N1 influenza virus, five of which were downregulated, 23 miRNAs were upregulated and 71 miRNAs were expressed similar level compared to uninfected control Venn-Euler diagram shows a summary of differentially expressed miRNAs in response to influenza A virus infection. (a) Upregulated miRNAs when infected with pH1N1, H5N1 and H3N2. (b) Downregulated miRNAs when the cells were infected by pH1N1, H3N2, and H5N1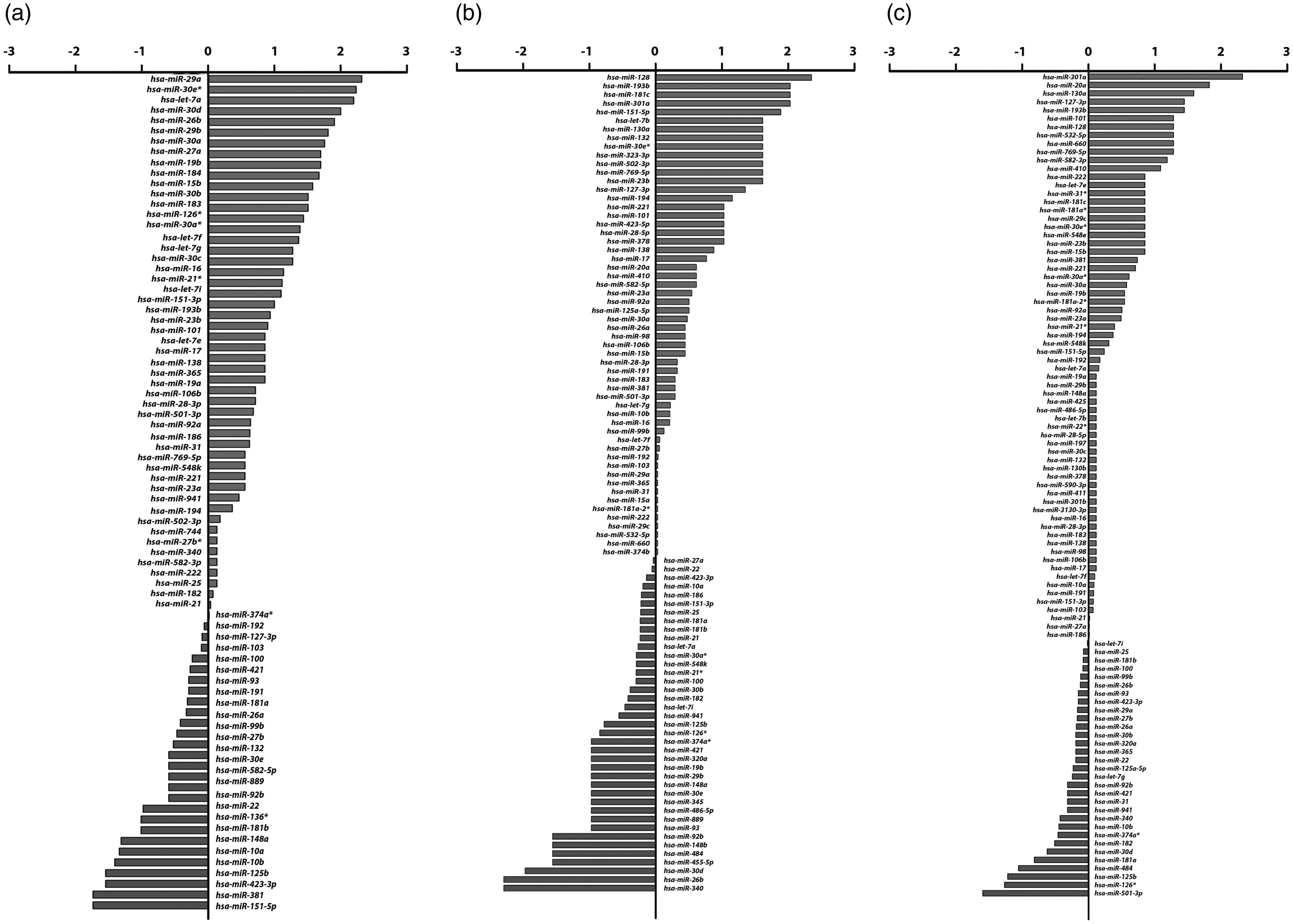
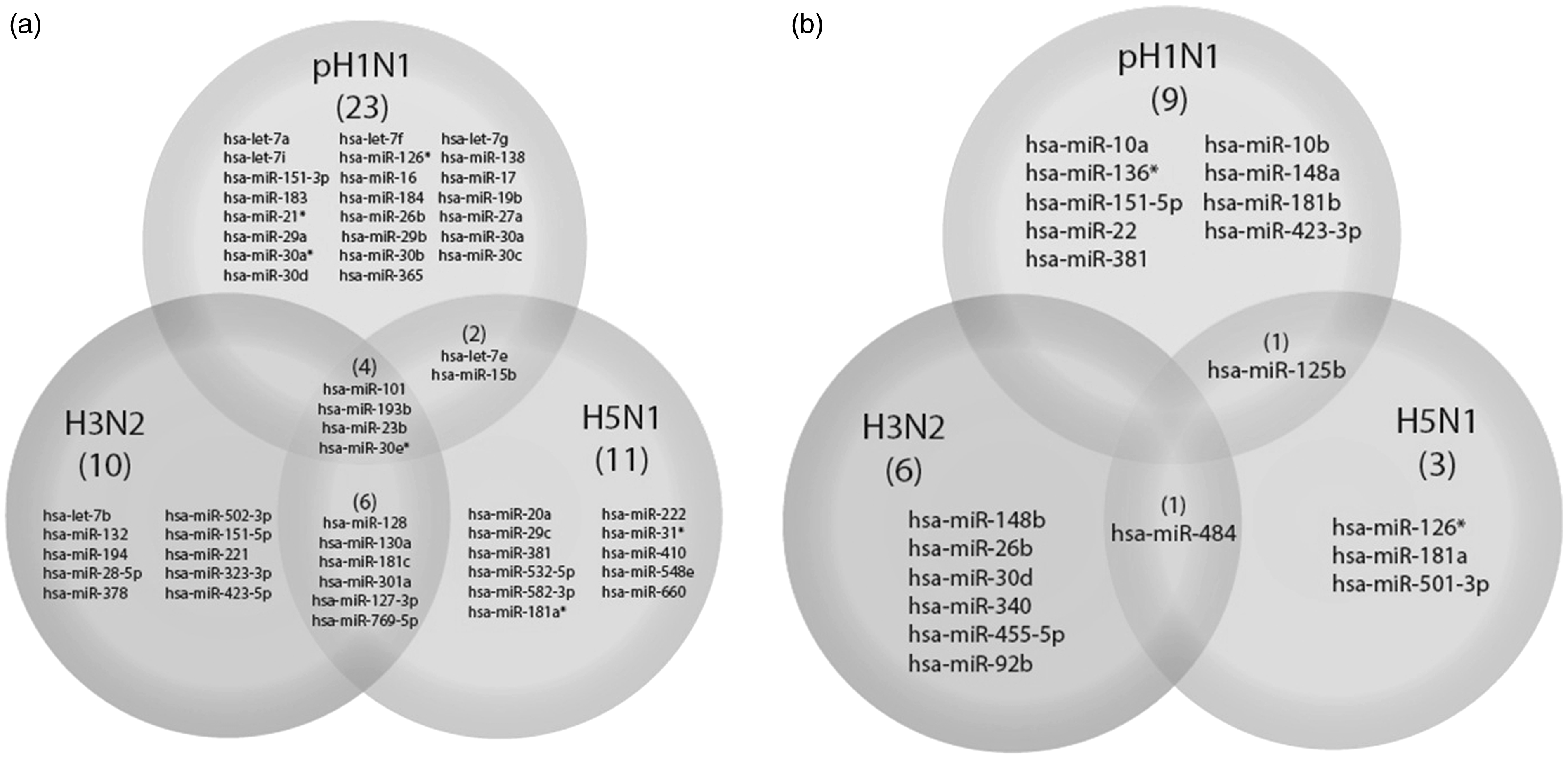
Analysis of common miRNA profiles in response to influenza A virus infection
Common miRNAs expression profiles in response to influenza A virus infection obtained from several studies
Host target genes prediction of miRNA expression profile
Numbers of validated target genes of common miRNAs in response to influenza A virus
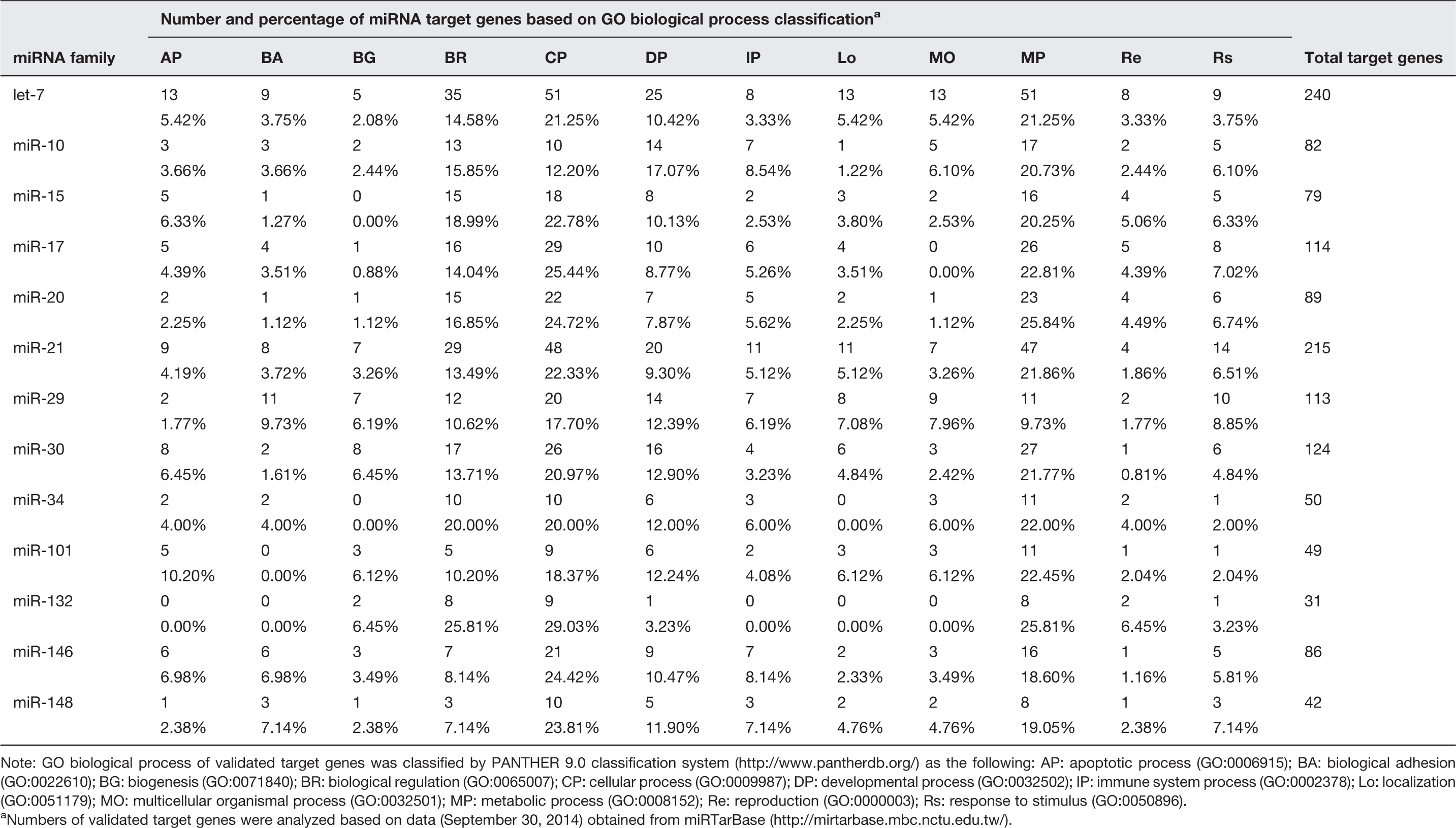
Note: GO biological process of validated target genes was classified by PANTHER 9.0 classification system (http://www.pantherdb.org/) as the following: AP: apoptotic process (GO:0006915); BA: biological adhesion (GO:0022610); BG: biogenesis (GO:0071840); BR: biological regulation (GO:0065007); CP: cellular process (GO:0009987); DP: developmental process (GO:0032502); IP: immune system process (GO:0002378); Lo: localization (GO:0051179); MO: multicellular organismal process (GO:0032501); MP: metabolic process (GO:0008152); Re: reproduction (GO:0000003); Rs: response to stimulus (GO:0050896).
Numbers of validated target genes were analyzed based on data (September 30, 2014) obtained from miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/).
The protein–protein interactions provide information regarding most biological processes in an organism. In this study, the prediction of target proteins interaction via four deregulated miRNAs from our experiment, and therefore miR-101, miR-193b, miR-23b, and miR30a, were illustrated. The analysis of topology demonstrates important network illustrations created by interacting proteins. Therefore, the interactions between proteins from the STRING 9.1 database were used to form the target gene network of the differentially expressed microRNAs to determine several ‘hub’ nodes (Figure 3), which play an important role in apoptosis during the infection of influenza viruses. This finding may enlighten the understanding of the promising roles of the differentially expressed microRNAs.
MicroRNA regulated network of target genes related to apoptosis in response to influenza virus infection. The STRING 9.1, together with data from Panther 9.0 constructed the interactions between proteins playing an important role in programmed cell death. As integrated with in silico analysis of microRNA target prediction, the pathway revealed the differentially expressed microRNAs controlling numerous target genes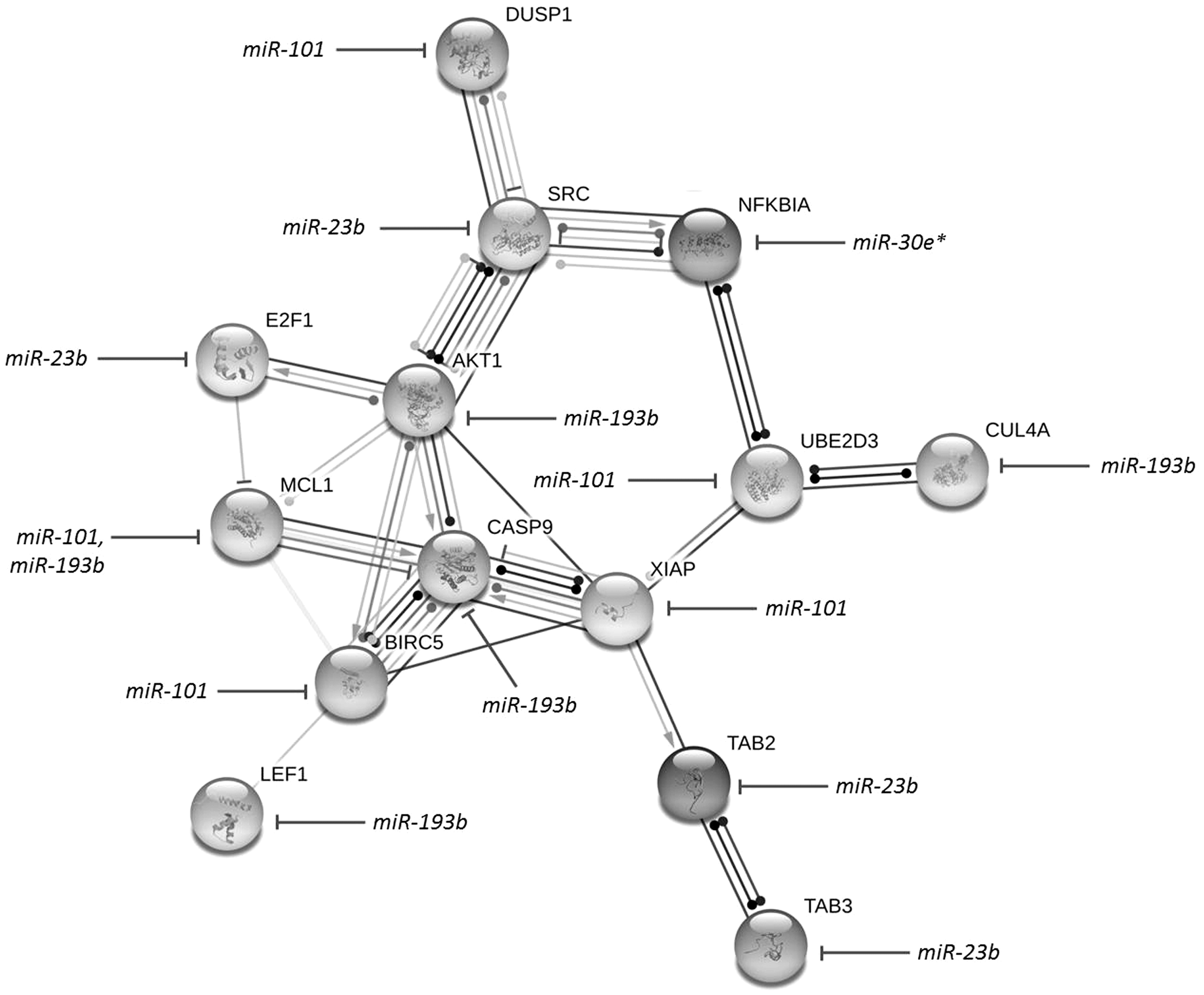
Viral target genes prediction of miRNAs
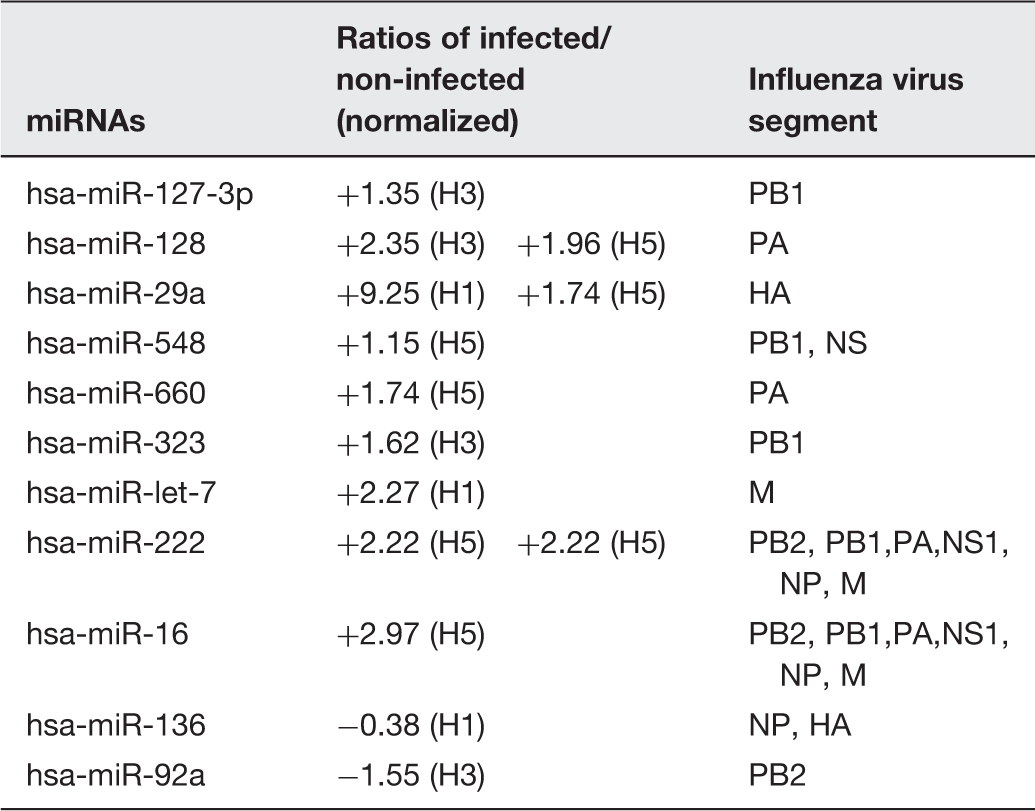
Viral targets of differentially expressed miRNAs in A549 infected with 3 subtypes of influenza virus
Discussion
The present study provided miRNA profiling at an early stage of infection with three subtypes of influenza A virus (pH1N1, H3N2 and H5N1) by using next-generation sequencing. From the miRNA profiling results it was found that a number of host genes could be regulated by influenza A virus via the miRNA mechanism. The differential miRNA expressions can reveal the host-pathogen interaction in viral infection in two ways. First, the virus can hijack the host cellular mechanism by up- or downregulating the miRNAs involved in pathways that can promote viral replication. For example, the results showed that miR-181c was found to be upregulated in cells infected with H5N1 and H3N2 influenza A virus. The increase of this miRNA can target many genes associated with cellular immune defense, such as BCL2, IL-2, and Tnf,49–51 and the deregulation of target genes can facilitate the influenza virus to avoid the suppressed host immune system. Second, the host cell can respond to viral infection via the miRNA mechanism by alteration of miRNAs which can have the effects on viral replication process to limit viral propagation. For example, the upregulation of miR-301 in H5N1 and H3N2 virus infection which can inhibit NKRF, the NFKB repressing factor, will promote the cytokine which indicates the innate and adaptive immunity response in mammalian cells. 52
Many previous studies have suggested a correlation between influenza A virus infection and miRNA alteration. The infection of influenza virus can specifically activate the alteration of miRNAs including let-7 family, miR-30a-c, miR-132 and miR-30e* which might target in many genes function in lung repair.44,53 However, the previous study found that miRNAs in let-7 family (let-7c, -7f, -7i), miR-30b and miR-26b were also upregulated by respiratory syncytial virus (RSV) infection, 53 implying that the alteration of these miRNAs are not only present against influenza virus infection but also other respiratory viruses.
The miRNA expression profiles were analyzed and compared to those from several studies which were obtained from different sample sources, viral strains, infection periods, and culture models, in order to discover the common miRNAs that overlapped between different systems. The results showed that some miRNAs were required to be considered. The miR-21, -29, -30, -34 and let-7 family were widely responsive to influenza virus infection in many systems. Interestingly, miR-15 and miR-21 were upregulated by influenza virus infection while miR-132, -30, and -101 were upregulated only in the human model system.
After the target prediction had been performed, most of the mechanisms due to virus infection were related to cellular processes, metabolic processes, developmental processes, and biological regulation (Table 2 and Table S1, Supplementary Information). Many miRNAs and their target genes should be considered and further discussed. MiR-101 can target MCL1, the anti-apoptosis gene and PRKDC, one of the large family of serine/threonine kinase, 54 which can play an important role in the receptor-mediated endocytosis process of the influenza life cycle, since the HA protein of the influenza virus can efficiently activate the PKC signaling pathway.55,56 It has been reported that the influenza virus propagation process can be suppressed by a specific inhibitor of PKC. 57 Another miRNA that should be considered is the regulation of cell apoptosis via miR-29c (Figure 3) which was found to be upregulated in H5N1 infection. Much evidence suggests that this miRNA is involved in the virus-induced apoptosis pathway via the repression of anti-apoptotic agent, BCL2L2. 41 This is related to a previous study that found that the miR-29 family tends to be more upregulated due to influenza virus infection. 58 Moreover, some of the miRNA experimentally targeted genes play a critical role in immunoregulatory functions. For example, the upregulation of miR-15b suggests that infection of influenza virus can suppress the host immune response via miRNA mechanism. A previous study found that the IL-1B gene, the crucial regulator of inflammatory response, cellular activity including cell proliferation, differentiation and apoptosis, can decrease the expression of miR-let-7g and miR-26b. 53 Corresponding to this study, the high expression of let-7g and miR-26b indicated that the IL-1B has not been produced in the first 24 h after infection. In addition, our study showed that the upregulation of let-7 family (in all 3 subtypes infection), miR-29a, b (in pH1N1 infection) and mir-29c (in H5N1 infection) might have resulted from TNF-α and IL-4 still being inactive in the early phase of infection. However, the downregulation of miR-26b by H3N2 influenza virus infection should be considered, as it might be responsible for the different pathogenicity of H3N2 virus from pH1N1 virus and RSV.
A number of miRNAs have been found to be down regulated. Most of them can target genes associated with DNA methylation, protein degradation, and the signal transduction pathway. For example, the downregulation of miR-125b can trigger the MAPK signaling pathway, which regulates various cellular responses such as cell proliferation and apoptosis.59,60 Previously, MAPK and CDK13 can be controlled by miR-548-3p and miR-138, which were, respectively, 61 identified as the mediators required for influenza virus replication. It is tempting to conclude that the upregulation of miR-548 (in H5N1 infection) and miR-138 (in pH1N1 infection) is the mechanism of the host response to limit influenza virus replication. Besides, a previous study suggested that the upregulation of MAPK protein can result from HA membrane accumulation due to the increase of polymerase activity. Interestingly, the upregulation of genes associated with DNA methylation via the decline of miRNA can cause the chromatin packaging, which can lead to the protein expression inhibition and may affect virus replication.
The difference of miRNA expression profile in cells infected with different subtypes might provide insight in the distinct pathogenicity of influenza virus. The result showed that there are four upregulated miRNAs including hsa-miR-101, hsa-miR-193b, hsa-miR-23b, and hsa-miR-30e*. Moreover, there were some strain-specific miRNAs which expressed in different levels in each subtype of influenza virus. The results showed that infection of H5N1 HPAI influenza virus can cause the highest amount of differential miRNA expression. Compared to a previous study, miR-31and miR-29c which are upregulated in H5N1 infection, and miR-29a and b, which are upregulated in pH1N1 infection, were confirmed to be potentially controlled during the early stage of influenza virus infection. 38 Interestingly, the results showed that miR-484 was found to be deregulated in cells infected with H5N1 and H3N2 influenza A virus. This miRNA can inhibit furin, the proteolytic enzyme that can cleave HA0 protein to facilitate influenza virus entry step. The downregulation of miR-484 will allow the host cell to produce more furin and promote the furin-dependent entry mechanism of the influenza viral life cycle.62–64 One point of discussion should be considered, infection of the pH1N1 virus can recruit the miR-30 family, including miR-30a, miR-30a*, miR-30b, and miR-30c while only one member of miR-30 family was found to be deregulated in H3N2 infection, contrary to a previous study. 11 However, the previous study suggested that genetics of the subject might account for the regulation of miRNA expression, 65 so that genetic variation and a different cell culture system can affect the miRNA expression during influenza virus infection. While the previous study found that the change of miR-21*, miR-100*, miR-141, miR-574-3p, miR-1274a and b resulted from H5N1 infection in NCI-H292 cell line, 12 our results show the upregulation of miR-21* only. This suggested that the different cell culture might be responsible for various cellular responses via the miRNA mechanism. Moreover, compared to a previous study, 66 which investigated the miRNA profile of A/Udorn/72 (H3N2) infection, and suggested that the miRNA profile of the same subtype of influenza can be altered depending on the different genetic characteristics of the virus, the up-to-date virus infection can represent the virus that is presently circulating. In the study of Zhao et al., 45 infection of canine influenza virus in lungs and trachea of dogs can cause the abundant expression of miR-143 and let-7. When compared to our study, the difference of miR-143 expression level may be due to the different characteristics of viral strains or host cells.
Infection with virus can alter many cellular mechanisms such as apoptosis which is one of the main machinery. Herein, we identified the genes and miRNAs observed in high-throughput sequencing that are involved in the apoptotic pathway by using STRING software (Figure 3). The pathway revealed the differentially expressed miRNAs controlling numerous target genes including NFКB, SOC, STAT, and IFN which can be regulated by many interesting miRNAs, including let-7 family, miR-101, miR-30, and miR-21, which might lead to development of miRNA therapeutics for influenza-like illness. However, many further experiments should be required such as western blot, in vivo study and pull down assays to make the refined observation and development.
Most of the upregulated miRNAs were predicted to target the influenza virus genome (Table 3). Most of them could bind to genes that have been reported in the early stage of influenza virus life cycle such as hemagglutinin and polymerase (PB2, PB1 and PA), which play the vital role in influenza virus entry mechanism and genome replication, respectively. In addition, a previous study suggested that miR-548an can mediate the influenza virus infection by regulation of non-structural-1A binding protein (NS1ABP), 42 corresponding to our study which found that the miR-548 was upregulated in H5N1 infection, implying that the host cell can produce more miR-548 against the exposure of influenza virus from an early stage of infection. Some miRNAs were found to be downregulated, suggesting that the virus can also trigger the miRNA pathway to fine-tune the miRNA mechanism to survive in host cells. The study in other time points after virus infection might reveal other differential miRNAs that can target other sites of the influenza virus genome, which may lead to the insight into the cellular mechanisms used to fight back against influenza virus infection. However, the built-on study by using widely used panel of robust tools, which are specific miRNAs inhibitors and miRNA mimics to knock down or increase the expression of miRNAs, is crucial to investigate the specific role of these candidate miRNAs.
Conclusion
In summary, this study provides the overall analysis of miRNA profiles in cell cultures infected with three different subtypes of influenza virus which have caused sporadic outbreaks in the human population, to gain insight into the overview of how the influenza virus can trigger the host cellular mechanism to facilitate itself to survive in host cells and also to find how cells can manipulate the cellular process to defend against influenza virus infection via the microRNA mechanism. This study provides information only in the early stage of infection, at 24 h post infection and can generate some potential candidate miRNAs and predicted target genes. This information can lead to the development of effective therapeutic agents, which have to interfere or control the influenza life cycle at an early stage of infection. Moreover, to explore the novel antiviral drugs, further analysis will be required such as miRNA profiling in late stages of infection or the study of knock-down and overexpression of candidate miRNAs.
Footnotes
Authors’ contributions
JM analyzed the miRNA profiling and drafted the manuscript. WP carried out the NGS process, data processing and visualization. SS performed protein interaction network. KK assisted in cell culture and virus infection. KP assisted in NGS libraries preparation. TJ assisted in data analysis. YP provided stock of viruses and revised the manuscript. SP designed the study, data analysis, revised the manuscript and coordination. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to gratefully acknowledge the Department of Biochemistry and Research affairs, Faculty of Medicine, Chulalongkorn University for their invaluable contribution to the success of this study. Funding for this study was supported by the Thailand Research Fund (TRF: RSA5680031); the Postdoctoral Scholarship, Ratchadapiseksompotch Fund (Faculty of Medicine, Chulalongkorn University); National Research University Project; Office of Higher Education Commission (WCU-007-HR-57); Centenary Academic Development Project (CU56-HR01); the Ratchadaphiseksomphot Endowment Fund of Chulalongkorn University (RES560530093); Research Chair Grant, National Science and Technology Development Agency (NSTDA) and Chulalongkorn Academic Advancement into its 2nd Century Project.
Declaration of Conflicting Interests
The authors hereby declare no personal or professional conflicts of interest with any aspect of this study.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.