
Abstract
Macrophage colony-stimulating factor is a vital factor in maintaining the biological function of monocyte–macrophage lineage. It is expressed in many tumor tissues and cancer cells. Recent findings indicate that macrophage colony-stimulating factor might contribute to chemoresistance, but the precise mechanisms are unclear. This study was to explore the effect of macrophage colony-stimulating factor on doxorubicin resistance in MCF-7 breast cancer cells and the possible mechanism. In the study, the human breast cancer cells, MCF-7, were transfected with macrophage colony-stimulating factor. We document that cytoplasmic macrophage colony-stimulating factor induces doxorubicin resistance and inhibits apoptosis in MCF-7 cells. Further studies demonstrated that cytoplasmic macrophage colony-stimulating factor-mediated apoptosis inhibition was dependent on the activation of PI3K/Akt/Survivin pathway. More importantly, we found that macrophage colony-stimulating factor-induced autophagic cell death in doxorubicin-treated MCF-7 cells. Taken together, we show for the first time that macrophage colony-stimulating factor-induced doxorubicin resistance is associated with the changes in cell death response with defective apoptosis and promotion of autophagic cell death.
Introduction
Macrophage colony-stimulating factor (M-CSF), also known as colony-stimulating factor (CSF-1), can promote monocyte–macrophage cell growth, proliferation, and differentiation, as well as maintenance of the biological functions of monocyte–macrophage.1,2 In recent years, some studies show that M-CSF plays an important role in tumorigenesis, which has been verified in lymphoma, lung cancer, ovarian cancer, breast cancer, and HL-60 leukemia.3–7 And the nuclear staining of M-CSF indicated enhanced metastatic potential and poor prognosis in breast cancer cells. 8 Similarly, the high expression of cytoplasmic M-CSF in MDA-MB-231 breast cancer cells contributed to the invasion and metastatic of tumor in a mouse model. 9 On the other hand, M-CSF antibody can reverse the chemoresistance of human MCF-7 breast cancer xenografts, 10 which suggested that M-CSF might have a role in tumor chemoresistance.
Chemoresistance is a major barrier for the successful treatment of cancer, and defect in apoptosis underlies chemoresistance in most tumors. Apoptosis can be inhibited by various survival signaling mechanisms in cancer cells. One such mechanism is the activation of PI3K/Akt pathway, which inactivates Bad that weaken apoptosis.11,12 Interestingly, M-CSF can also activate PI3K/Akt pathway. 13 Thus, we speculate that the effects of M-CSF on chemoresistance may depend on PI3K/Akt pathway.
Autophagy is an evolutionarily conserved intracellular degradation process, and it plays an important role in tumor development and chemoresistance of cancer cells.14,15 For example, autophagy induction with RAD001 enhanced chemosensitivity through Met inhibition in papillary thyroid cancer. 16 In addition, autophagy is also associated with paclitaxel resistance in MCF-7 breast cancer cells. 17 Furthermore, the latest study showed that autophagy has a vital role in the biological function of M-CSF. For instance, autophagy was required for M-CSF-induced macrophagic differentiation. 18
Therefore, we propose that the effect of M-CSF on chemoresistance is possibly mediated by autophagy and apoptosis. In this study, we found that cytoplasmic M-CSF-induced doxorubicin (Adriamycin, ADM) resistance is mediated by apoptosis inhibition through activation of the PI3K/Akt/Survivin pathway in MCF-7 cells. Importantly, M-CSF induce autophagic cell death in MCF-7 cells under doxorubicin treatment. Thus, we postulate that the switch from apoptotic to autophagic cell death, at least in part, is responsible for chemoresistance in MCF-7 breast cancer cells.
Materials and methods
Cell lines and reagents
MCF-7, a human breast cancer cell line, was obtained from ATCC (Manassas, VA). The MCF-7-M cells were transfected with M-CSF in MCF-7 cells. The MCF-7-C cells were transfected a control plasmid (empty vector) in MCF-7 cells. MCF-7, MCF-7-C, and MCF-7-M cells were maintained in RPMI 1640 (GIBCO BRL, Grand Island, NY) supplemented with 10% FBS and antibiotics at 37℃ with 95% air and 5% CO2.
Primary antibodies against Akt, p-Akt (S473), PI3K were purchased from Epitomics (Burlingame, CA). Primary antibodies against Survivin, LC3, and β-actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Primary antibodies against p-PI3K (P85α) were from Bioword (Louis Park, MN). The horseradish peroxidase (HRP)-conjugated anti-rabbit IgG, anti-goat, and anti-mouse IgG were from Beyotime (Haimen, China).
LY294002 (PI3K inhibitor) was purchased from Beyotime (Haimen, China). SH-6 (Akt inhibitor), YM155 (Survivin inhibitor), and RAD001 (an autophagy activator) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). 3-methyladenine (3-MA, an autophagy inhibitor) and doxorubicin were from Sigma (St Louis, MO).
Stable transfection
The cells were seeded into six-well plates at 7.5 × 104 cells per 500 µl per well in the 1640 containing 10% FBS for 24 h. Then, the cells were stably transfected with either pCMV/cyto/myc-M-CSF (Cytoplasmic M-CSF gene overexpressed) or pCMV/cyto/myc vector (Empty vector) using Lipofectamine 2000 reagent (Invitrogen, USA), as described by the manufacturer. After 6 h, fresh medium was added to the plates. After two days, the medium was replaced with the growth medium with selection reagent, G418 (500 µg/ml, Invitrogen, USA). Selection was continued for 15 days, with the medium refreshed every three days. In order to confirm the efficiency of stable transfection, the M-CSF expression was determined by Western blot analysis in MCF-7 cells.
Western blotting analysis
Cells were washed with cold PBS and mechanically homogenized in RIPA lysis buffer (Beyotime, China). Total protein samples (60 µg/well) were separated on 10% or 15% SDS-PAGE gels and transferred to PVDF membranes (Millipore, USA). After washing for 10 min in TBST (0.1% Tween-20, TBS) three times, the membranes were placed into blocking buffer (5% non-fat milk in TBST) for 1 h at room temperature. The membranes were then incubated with primary antibodies against PI3K (1:2000), p-PI3K (1:800), Akt (1:10000), p-Akt (1:2000), Survivin (1:400), LC3 (1:400), and β-actin (1:2000) overnight at 4℃. The next day, after washing for 15 min in TBST (0.1% Tween-20, TBS) four times, the membranes were incubated with goat anti-rabbit IgG-HRP (1:4000), goat anti-mouse IgG-HRP (1:4000), rabbit anti-goat IgG-HRP (1:4000) for 1 h at 37℃ and visualized by ECL-PLUS reagents (Beyotime, China). β-actin was used for normalization of protein expression.
Enzyme-linked immunosorbent assay for M-CSF assay
The cell supernatants were collected and analyzed using the enzyme-linked immunosorbent assay (ELISA) for the secretion level of M-CSF. Analysis was performed in line with the manufacturer’s protocol for a commercial kit (Invitrogen Corporation).
Cell viability assay
Cells were plated in 96-well plates at 1 × 104 cells per 100 µl per well in the 1640 containing 10% FBS. After 16 h, medium was replaced with the same medium containing (0, 0.5, 1, 2, 4, and 8 µmol/l doxorubicin with or without other reagents). After 24 h, 20 µl/well of MTT (5 mg/ml) (Solarbio, China) was added. Then cells were incubated for 4 h at 37℃. Afterwards, the supernatant was removed and the formazan precipitates were dissolved with 150 µl DMSO. The absorbance was measured at 570 nm by the use of an ELX-800 microplate assay reader (Elx800, Bio-tek, USA).
Heochst 33342 staining for apoptosis assay
Cells were plated in 24-well plates (5 × 104 cells/well in 1 ml) in the 1640 containing 10% FBS. 12 h later, the medium was replaced with the serum-free medium. 24 h later, the serum-free medium was replaced with the same medium containing doxorubicin (0, 2 µmol/l) with or without other reagents. After 24 h drug incubation, the medium was removed and Immunol Staining Fix Solution (Beyotime, China) was added at 0.5 ml per well for 20 min at 4℃. Then, the plates were washed two times for 3 min in PBS. After washing, Heochst 33342 staining fluid (Beyotime, China) was added at 0.5 ml per well and incubated for 20 min at 37℃. Afterwards, the plates were washed two times for 3 min in PBS and visualized by a fluorescence microscope (Olympus IX 70, Japan). The characteristics of apoptotic cells are chromatin condensation and fragmentation in nucleus.
Annexin V-fluorescein isothiocyanate apoptosis assay
An annexin V-FLUO Staining Kit (Boehringer-Mannheim, Germany) was used to evaluate doxorubicin-induced apoptosis. Cells were cultured in a six-well plate and exposed to doxorubicin with or without other reagents. The cells were harvested and stained with annexin V-FLUOS and PI for 15 min. Apoptosis was immediately analyzed with a flow cytometer (Beckman Coulter, Fullerton, CA) at the wavelength of 488 nm.
Statistical analysis
All results were expressed as mean ± SD. Data analysis was performed using SPSS 18.0 (SPSS, Chicago, IL). Groups were compared using Student’s t-test or one-way ANOVA. Differences were deemed statistically significant at P < 0.05.
Results
M-CSF expression was upregulated in overexpressed transfectants of MCF-7 cells
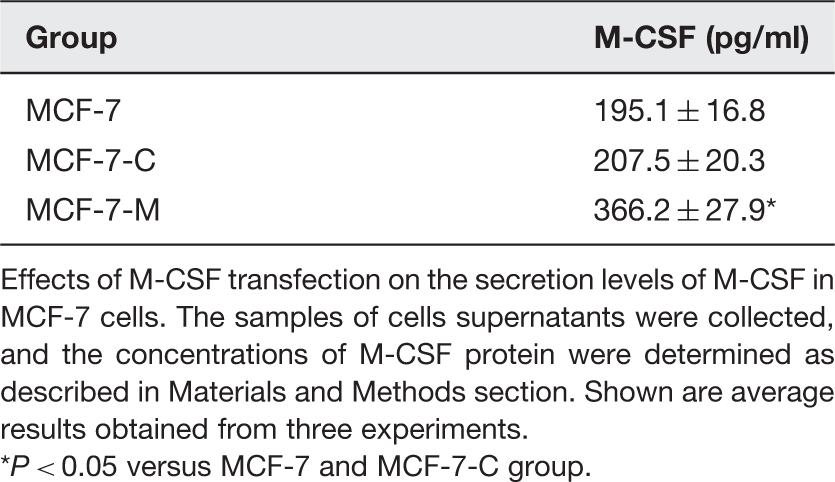
To determine the efficiency of M-CSF stable transfection, Western blotting was performed to analyze protein expression in the MCF-7, MCF-7-C, and MCF-7-M cells (Figure 1(a) and (b)). We found that the expression of M-CSF in MCF-7-C cells was similar to MCF-7 cells. Compared to the MCF-7 and MCF-7-C, a much higher expression of M-CSF protein was detected in MCF-7-M cells. Additionally, the secretion levels of M-CSF were tested by ELISA in MCF-7 cells. Compared with the controls (MCF-7 and MCF-7-C cells), the secretion levels of M-CSF were increased in the MCF-7-M cells (Table 1). The data showed that the MCF-7 cells were stably transfected with M-CSF.
M-CSF induces doxorubicin resistance in MCF-7 cells. (a) M-CSF protein expression by Western blot in MCF-7, MCF-7-C, and MCF-7-M cells. (b) Band densitometry analysis of M-CSF expression in MCF-7, MCF-7-C, and MCF-7-M cells normalized to β-actin, respectively. (C) Cell survival rate analysis by MTT in doxorubicin-treated MCF-7, MCF-7-C, MCF-7-M, and MCF-7-M cells incubated with DMSO, LY294002, SH-6, or YM155. The groups of DMSO1 and DMSO2, respectively, compared with the groups of LY294002 and SH-6, because the DMSO dosage was different in dissolution of LY294002 and SH-6. Results represent mean ± SD (*P < 0.01). (A color version of this figure is available in the online journal.) Analysis of the secretion levels of M-CSF in MCF-7 cells Effects of M-CSF transfection on the secretion levels of M-CSF in MCF-7 cells. The samples of cells supernatants were collected, and the concentrations of M-CSF protein were determined as described in Materials and Methods section. Shown are average results obtained from three experiments. *P < 0.05 versus MCF-7 and MCF-7-C group.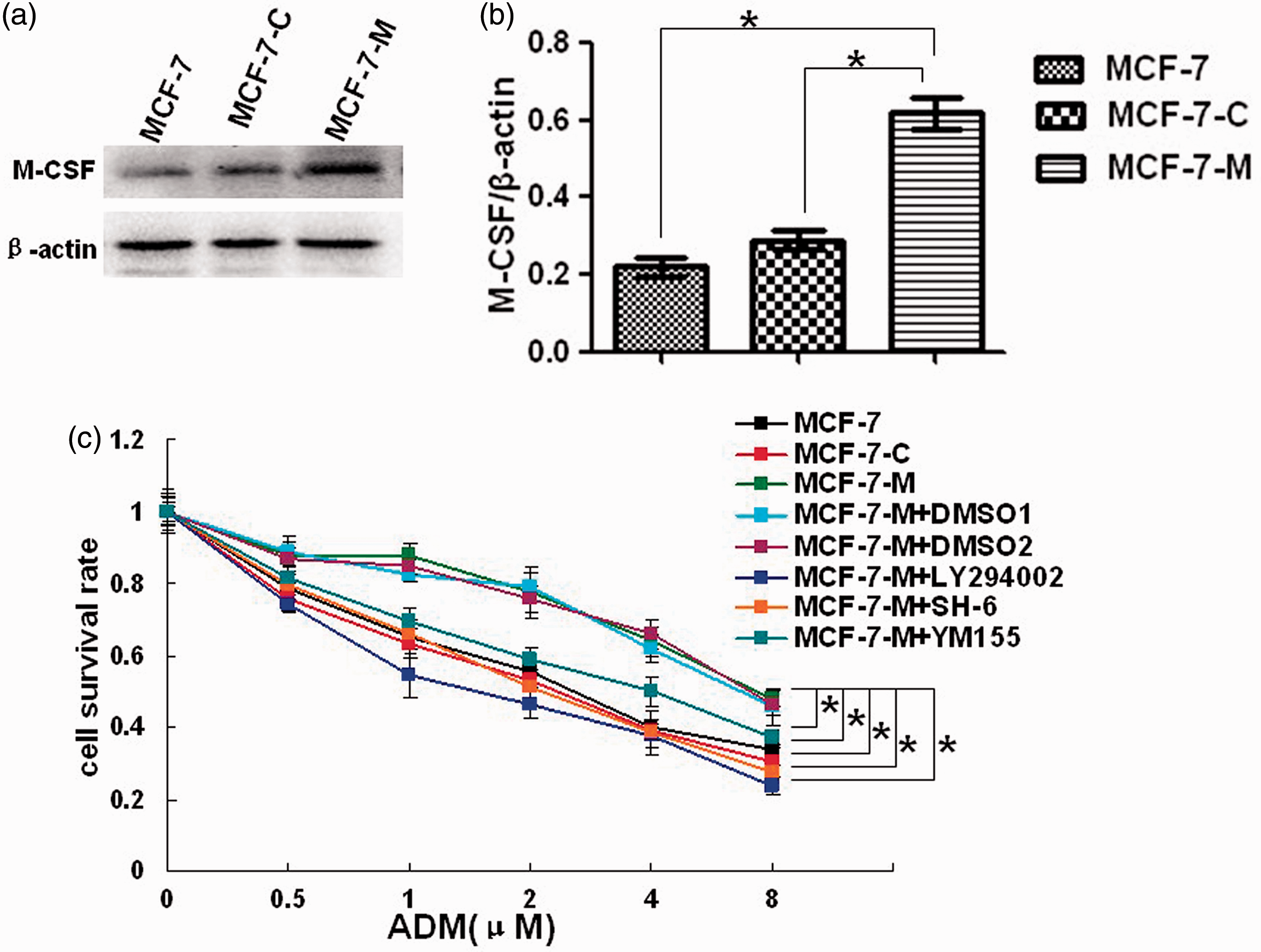
M-CSF-induced doxorubicin resistance in MCF-7 cells
As mentioned earlier, M-CSF antibody can reverse the chemoresistance of human MCF-7 breast cancer xenografts. 10 Thus, we speculate that M-CSF may induce chemoresistance of MCF-7 cells. To determine whether M-CSF influences the chemosensitivity of MCF-7 cells, we compared the effect of doxorubicin on MCF-7-M, MCF-7, and MCF-7-C cells by a 24-h MTT assay (Figure 1(c)). As expected, MCF-7-M cells were more resistant to the cytotoxic effects of doxorubicin compared with MCF-7 and MCF-7-C cells.
M-CSF blocked chemotherapy-induced apoptosis in MCF-7 cell
Acquired chemoresistance is one of the major challenges in patients with advanced stage malignancies. Studies have revealed that a lot of factors promote chemoresistance in cancer cells. Such factors include increased drug efflux, enhanced DNA damage repair, high intracellular ATP levels, and defects in apoptosis.19,20 Apoptosis, which can be inhibited by PI3K/Akt pathway, is an important aspect of chemotherapy-induced cell death. Additionally, it is interesting that M-CSF can activate PI3K/Akt pathway through binding of its receptor.
13
So, we speculate that M-CSF may inhibit chemotherapy-induced apoptosis in MCF-7 cells. To test this, we measured apoptosis in MCF-7, MCF-7-C, and MCF-7-M cells, which were incubated with doxorubicin at different concentration for 24 h and stained by Heochst 33342 staining in 24-well plates. Based on nuclear morphological alterations visualized by a fluorescence microscope, we observed a significant reduction of doxorubicin-induced apoptosis in MCF-7-M cells compared with MCF-7 and MCF-7-C cells (Figure 2(a) and (b)). Consistent with Heochst 33342 staining, the apoptosis rate analyzed by flow cytometry was low in MCF-7-M cells, which exhibited a fivefold decrease in the percentage of annexin V-positive cells compared with MCF-7 and MCF-7-C cells (Figure 2(c) and (d)). These results suggested that M-CSF induce doxorubicin resistance through inhibition of apoptosis in MCF-7 cells.
M-CSF-induced apoptosis resistance in doxorubicin-treated MCF-7 cells is dependent on PI3K/Akt/Survivin pathway. ((a) and (b)) Apoptosis analysis by Heochst 33342 staining in 0.5 µM (a) or 2 µM (b) doxorubicin-treated MCF-7, MCF-7-C, MCF-7-M, and MCF-7-M cells incubated with DMSO, LY294002, or YM155. (c) Apoptosis analysis by flow cytometric in 0.5 µM doxorubicin treated MCF-7, MCF-7-C, MCF-7-M, and MCF-7-M cells incubated with LY294002 or YM155. (d) Band densitometry analysis of apoptosis rate which represented in (c). Results represent mean ± SD (*P < 0.01). (A color version of this figure is available in the online journal.)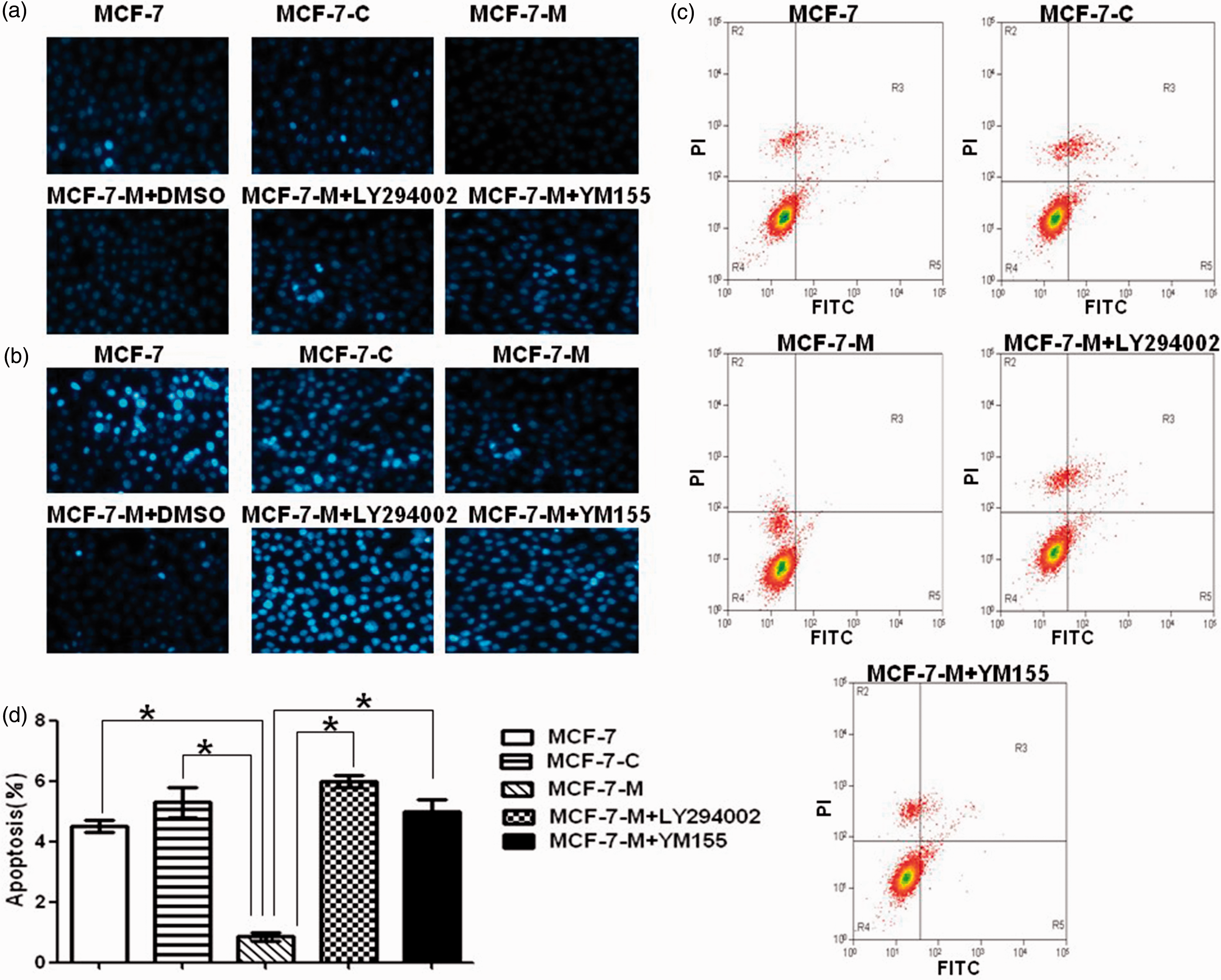
M-CSF-induced doxorubicin resistance was mediated by apoptosis inhibition through activation of PI3K/Akt/Survivin pathway in MCF-7 cells
Survivin is an important member of inhibitors of apoptosis proteins (IAPs) and is expressed in majority of breast cancers.
21
Similar to other IAPs, Survivin has the capacity to inhibit pro-apoptosis factors such as caspase-3 and caspase-7 and its overexpression leads to resistance of apoptosis caused by chemotherapy in cancer cells.22,23 Thus, Survivin may play a crucial role in chemoresistance of cancer cells. In our study, we determined the protein expression of Survivin in MCF-7, MCF-C, and MCF-7-M cells. Consistent with the speculation mentioned above, we found increased Survivin expression in MCF-7-M cells that were resistant to doxorubicin treatment compared to MCF-7 and MCF-7-C cells (Figure 3(a)). These results suggested that M-CSF-induced doxorubicin resistance may be mediated by Survivin overexpression.
M-CSF activates PI3K/Akt/Survivin pathway and induces autophagy in MCF-7 cells. (a) Survivin protein expression by Western blot in MCF-7, MCF-7-C, and MCF-7-M cells. (b) Western blots of PI3K, phosphorylated PI3K, Akt, phosphorylated Akt, and Survivin expression in MCF-7, MCF-7-C, MCF-7-M, and MCF-7-M cells incubated with DMSO or LY294002. (c) LC3II protein expression by Western blot in doxorubicin treated or not treated MCF-7, MCF-7-C, and MCF-7-M cells. (d) MCF-7-M cells were treated with doxorubicin, and then LC3II protein expression was analysis by Western blot at the indicated time points and concentration points. Results represent mean ± SD (*P < 0.01, #P < 0.05)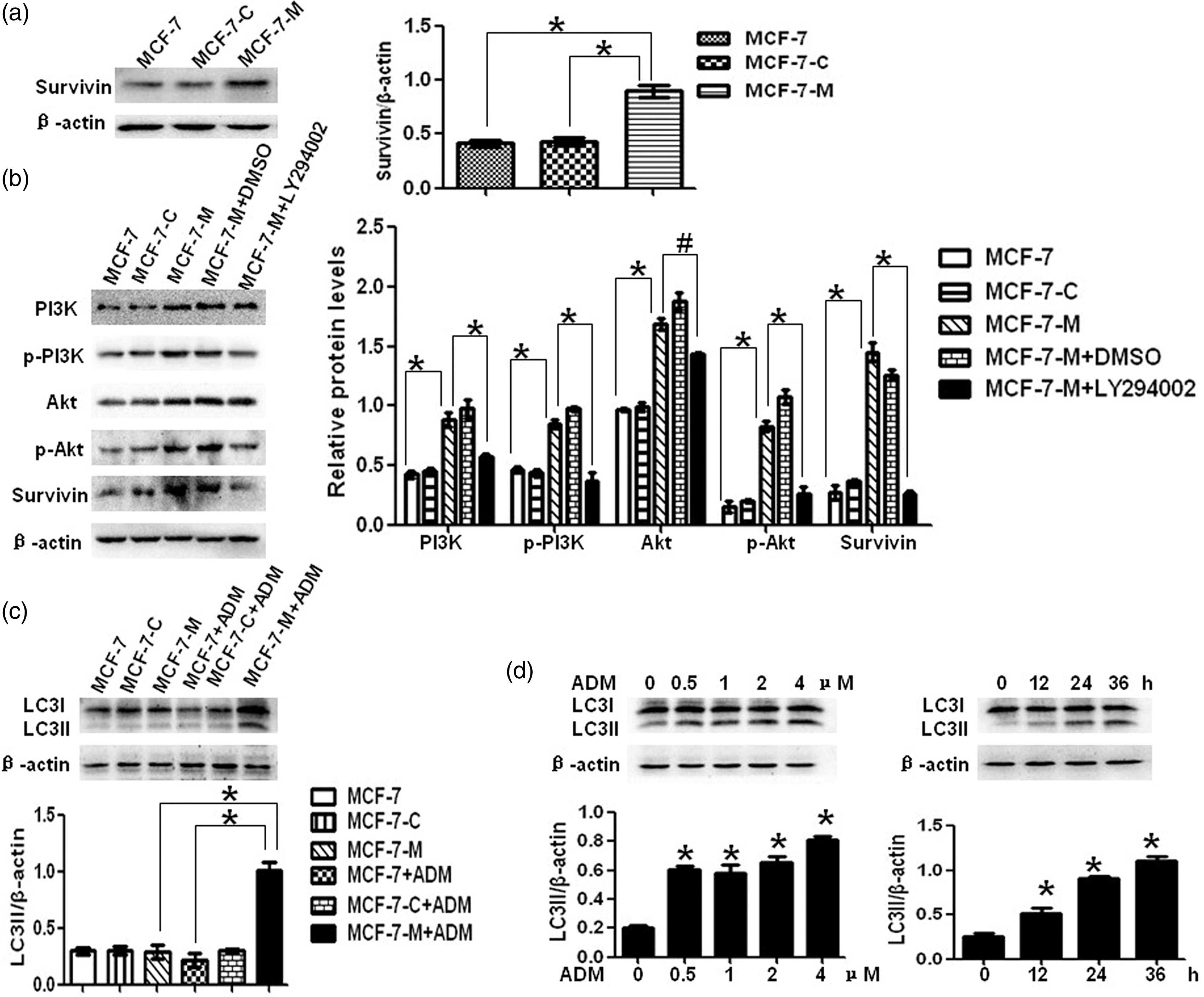
To determine the exact mechanism of Survivin overexpression, we analyzed the activation of PI3K/Akt pathway, which plays an important role in IAPs expression through Western blot. As expected, PI3K/Akt pathway was hyper-activated in MCF-7-M cells with demonstrable increase in protein expression of PI3K, phosphorylated PI3K, Akt, and phosphorylated Akt when compared with MCF-7 and MCF-7-C cells (Figure 3(b)). Further, the PI3K inhibitor LY294002 and also the downstream Akt inhibitor SH-6 were used to determine whether PI3K/Akt activation was responsible for Survivin overexpression. The expression of Survivin was inhibited by either LY294002 or SH-6 (Figure 3(b) and Figure S1). It was suggested that M-CSF-induced Survivin overexpression was dependent on PI3K/Akt pathway.
Furthermore, we tested the effect of M-CSF-induced PI3K/Akt/Survivin pathway on chemoresistance of MCF-7 cells through cell viability assay. Blocking PI3K/Akt/Survivin pathway with LY294002, SH-6, or Survivin inhibitor YM155 significantly decreased doxorubicin resistance of MCF-7-M cells (Figure 1(c)). In addition, further prove the functional role of M-CSF-induced PI3K/Akt/Survivin pathway on apoptosis inhibition; we examined apoptosis in doxorubicin treated MCF-7-M cells that were incubated with or without LY294002 or YM155. Consistently, doxorubicin-induced apoptosis was markedly increased in LY294002 or YM155 incubated MCF-7-M cells (Figure 2). Taken together, our results indicated that M-CSF-induced doxorubicin resistance is mediated by apoptosis inhibition through activation of PI3K/Akt/Survivin pathway in MCF-7 cells.
M-CSF-induced doxorubicin resistance was associated with diminished apoptotic response but upregulated autophagic cell death
As described above, chemotherapy-induced apoptosis was inhibited by M-CSF through PI3K/Akt/Survivin pathway in MCF-7 cells. So, we consider that chemotherapy may induce autophagic cell death in apoptosis-deficient MCF-7-M cells. According to the guidelines for the use and interpretation of assays for monitoring autophagy, the increase of LC3II protein level is a more accurate indicator of autophagy. We first analyzed the autophagy level in doxorubicin-treated MCF-7, MCF-7-C, and MCF-7-M cells through LC3II protein expression. As shown in Figure 3(c), doxorubicin treatment induced a significant increase in LC3II expression in MCF-7-M cells but not in MCF-7 and MCF-7-C cells. In addition, we also found that doxorubicin treatment induced LC3II expression in MCF-7-M cells in a concentration and time-dependent manner (Figure 3(d)).
To determine whether autophagy induced by doxorubicin was lethal in MCF-7-M cells, we analyzed cell survival rate of doxorubicin treated MCF-7-M cells incubated with or without 3-MA. 3-MA was found to have significant cytotoxicity at concentrations higher than 2.5 mM in MCF-7-M cells (data not shown). Hence, 2.5 mM 3-MA was used for further studies. As shown in Figure 4(a), 3-MA not only inhibited doxorubicin-induced LC3II expression but also decreased the cytotoxicity of doxorubicin in MCF-7-M cells. Furthermore, RAD001 can enhance chemosensitivity to doxorubicin of MCF-7-M cells via active autophagy (Figure 4(b)). Taken together, these data suggested that doxorubicin could induce autophagic cell death in apoptosis-deficient MCF-7-M cells.
M-CSF induces autophagic cell death in doxorubicin-treated MCF-7 cells. (a) LC3II protein expression by Western blot in MCF-7-M and MCF-7-M cells treated with doxorubicin or doxorubicin and 3-methyladenine (3-MA). Cell survival rate analysis by MTT in doxorubicin-treated MCF-7-M cells which incubated with or without 3-MA. (B) Western blots of LC3II expression in MCF-7-M cells treated with RAD001 (0, 5, 10, 20, 40, and 80 nM) for 24 h. Cell survival rate analysis by MTT in doxorubicin-treated MCF-7-M cells which incubated with or without RAD001. Results represent mean ± SD (*P < 0.01)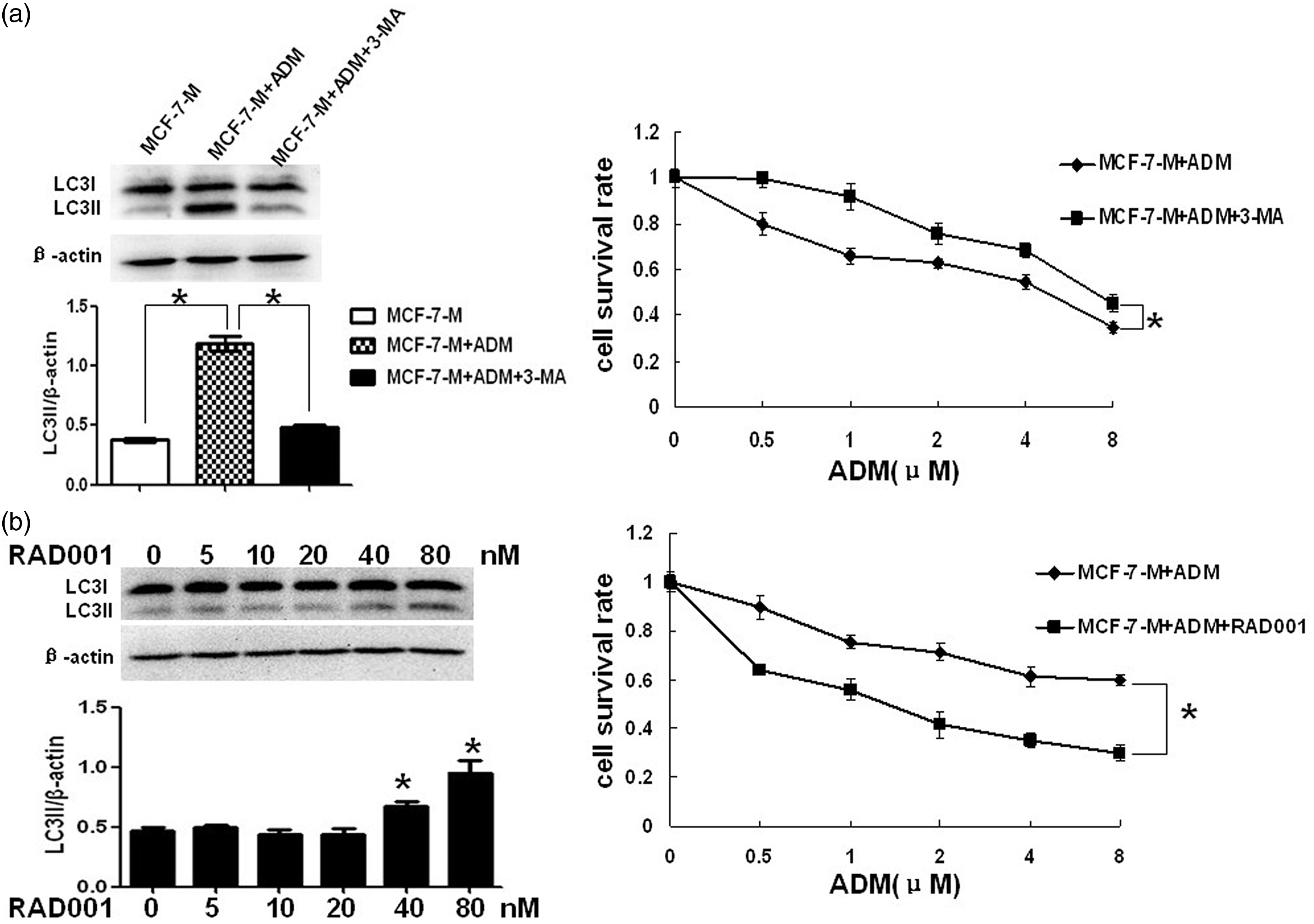
Discussion
In the current study, we demonstrated that cytoplasmic M-CSF inhibited apoptosis through activation of PI3K/Akt/Survivin pathway, subsequently leading to doxorubicin resistance of MCF-7 cells. Alternatively, cytoplasmic M-CSF-induced autophagic cell death in doxorubicin-treated MCF-7 cells. It was suggested that cytoplasmic M-CSF-induced doxorubicin resistance might be associated with switch from apoptosis to autophagic cell death in MCF-7 breast cancer cells.
M-CSF plays a fundamental role in maintaining the biological function of monocyte–macrophage and is expressed in many tumor tissue and cancer cells. 24 Role of M-CSF in tumorigenesis is attractive. Aharinejad et al. 9 discovered that overexpression of cytoplasmic M-CSF was responsible for the invasion and metastatic of cancer cell in a mouse model. In addition, a recent study showed that M-CSF antibody reversed the chemoresistance of human MCF-7 breast cancer xenografts. 10 These results suggested that M-CSF might act as a factor, which induce chemoresistance in MCF-7 breast cancer xenografts. But, the exact mechanisms of M-CSF in chemoresistance are not clear. Hence, we evaluated the specific effects of M-CSF on cancer cells by stable transfection of cytoplasmic M-CSF into MCF-7 cells. Our study demonstrated that cytoplasmic M-CSF-induced doxorubicin resistance in MCF-7 cells via changing in cell death responses through a switch from apoptosis to autophagic cell death as the principal mechanism of drug-induced cytotoxicity.
Apoptosis, which consists of extrinsic and intrinsic pathway, is the main cell death response to chemotherapy. Despite significant improvements in operative treatment, chemotherapy also acts as an important therapeutic strategy in patients with advanced stage malignancies. However, acquired chemoresistance is one of the major impediments to the successful treatment of cancer. Furthermore, defects in apoptosis are the common cause of chemoresistance. 25 In addition, apoptosis is dependent on the balance between apoptosis protein and IAPs in cancer cells. Survivin is an important member of IAPs and is expressed in majority of breast cancers. 21 Hence, we analyzed the effects of M-CSF on apoptosis in MCF-7 cells. We found that M-CSF-induced significant doxorubicin resistance of MCF-7 cell and the effect was mediated by apoptosis inhibition through activation of the PI3K/Akt/Survivin pathway. It was suggested that M-CSF-induced defects in apoptosis may be responsible for the doxorubicin resistance of MCF-7 cells. Since cell death is also found in MCF-7-M group, we speculated that there might be another cell death response to chemotherapy.
Autophagy is a programmed cell death that plays an important role in tumor development and chemoresistance of cancer cell. In our further study, we found that doxorubicin-induced autophagic cell death in MCF-7-M cells. This was consistent with recent findings describing the role of 5-Fluorouracil (5-FU) in inducing autophagic cell death in apoptosis-defective tumor cells. 26 In addition, rapamycin analogue RAD001 can increase chemosensitivity to doxorubicin in MCF-7-M via active autophagy. Tan et al. 27 also suggest that Src/STAT3-dependent heme oxygenase-1 induction mediates chemoresistance of breast cancer cells to doxorubicin by promoting autophagy.
Taken together, these results suggested that M-CSF-induced doxorubicin resistance involved diminished apoptotic response and upregulated autophagic cell death. This was consistent with the previous studies such as Ajabnoor et al., 17 who found that paclitaxel resistance was associated a switch from apoptotic to autophagic cell death in MCF-7 breast cancer cells. Another study showed cisplatin-resistant human clear cell carcinoma has greater sensitivity to RAD001 than parental cells. 28 These data indicated that cell death is predominantly via autophagy when the apoptotic response was diminished in the chemoresistance cancer cell. Even though the exact role of autophagy in chemoresistance is highly controversial, we are confident that M-CSF can induce autophagic cell death in MCF-7 cells under doxorubicin treatment. Considering autophagy has different roles in chemotherapy of cancer cells at different time periods, it may promote cell survival in early stages of chemotherapy, but inhibit cell survival at later stages. Autophagic cell death might be a modest cell death response to cancer cell chemotherapy. So, we determined that M-CSF-mediated doxorubicin resistance was associated with a switch from apoptosis to autophagic cell death in MCF-7 breast cancer cells. Although physiological and clinical implications of these effects induced by M-CSF in MCF-7 cells are yet to be determined in vivo, our study provides supportive evidence that M-CSF-induced doxorubicin resistance is associated with diminished apoptotic response and upregulated autophagic cell death in MCF-7 cells.
Footnotes
Author contributions
All authors participated in the design, interpretation of the studies and analysis of the data and review of the manuscript; MZ, XL, and ST contributed to the design of the study and performed all experimental procedures and statistical analysis and prepared the first draft of the manuscript. HZ, FT, YW, and ZM assisted in all experimental procedures and contributed to the writing of the manuscript.
Acknowledgments
This work was supported by Hunan School of Higher Learning Foundation of Science and 266 Technology Innovation Platform (09K072, 12K120), Youth Foundation of the Education Department of Hunan Province (11B110), Zhengxiang scholar program of the University of South China (Xiangyang Tang), and the Construct Program of The Key Discipline in Hunan Province (Basic Medicine Sciences in University of South China).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.