
Abstract
Fibroblast growth factor 21 (FGF21), a primarily liver-derived endocrine factor, has the beneficial effect of protecting blood vessels. Peroxisome proliferator-activated receptor γ (PPARγ), a ligand-activated nuclear transcription factor, has been reported to effectively inhibit pulmonary hypertension (PH). The purpose of this study is to investigate the role of FGF21 in hypoxia-induced PH (HPH) and explore the relationship between FGF21 and PPARγ in this disorder. Adult C57BL/6 mice were subjected to four weeks of hypoxia to establish a PH model. The effects of FGF21 and PPARγ agonists and antagonists were investigated in HPH mice, as well as the relationship between FGF21 and PPARγ in this model. Moreover, we investigated the underlying mechanisms of this relationship between FGF21 and PPARγ in vivo and in vitro. In vivo, we found that hypoxia resulted in pulmonary hypertension, right ventricular hypertrophy, pulmonary arterial remodeling, and pulmonary arterial collagen deposition. Furthermore, hypoxia decreased FGF21 and PPARγ levels. These changes were reversed by exogenous FGF21 and a PPARγ agonist and were further enhanced by a PPARγ antagonist. The hypoxia-induced decrease in β-klotho (KLB) expression was improved by the PPARγ agonist and further reduced by the PPARγ antagonist. Exogenous FGF21 increased adenosine monophosphate-activated protein kinase (AMPK) phosphorylation (Thr172) and PPARγ coactivator-1α (PGC-1α) expression in PH mouse lung homogenates. In vitro, we found that knockdown of AMPK or using an AMPK antagonist inhibited the FGF21-mediated up-regulation of PPARγ expression, and the PPARγ-mediated up-regulation of FGF21 expression was inhibited by knockdown of KLB. These results indicated that FGF21 exerts protective effects in inhibiting HPH. FGF21 and PPARγ mutually promote each other’s expression in HPH via the AMPK/PGC-1α pathway and KLB protein.
Impact statement
In this study, we reported for the first time that FGF21 alleviated hypoxia-induced pulmonary hypertension through attenuation of increased pulmonary arterial pressure, pulmonary arterial remodeling and collagen deposition in vivo, and we confirmed the mutual promotion of FGF21 and PPARγ in hypoxia-induced pulmonary hypertension. Additionally, we found that FGF21 and PPARγ mutually promote each other’s expression via the AMPK/PGC-1α pathway and KLB protein in vitro and in vivo. Pulmonary hypertension is a progressive and serious pathological phenomenon with a poor prognosis, and current therapies are highly limited. Our results provide novel insight into potential clinical therapies for pulmonary hypertension and establish the possibility of using this drug combination and potential dosage reductions in clinical settings.
Keywords
Introduction
Pulmonary hypertension (PH) is a fatal and progressive disease characterized by increased pulmonary arterial pressure (PAP) and pulmonary vascular resistance, which lead to right ventricular failure and even death.1,2 Pulmonary arterial remodeling, inflammation, endothelial dysfunction, aberrant pulmonary arterial smooth muscle cell (PASMC) proliferation, and collagen deposition are the major pathobiological features of PH that cause its pathogenesis and progression. 2 The exact mechanisms of this disorder remain unclear, and effective clinical treatments for PH are lacking. Pulmonary arterial collagen deposition plays a crucial role in hypoxia-induced pulmonary arterial remodeling and right ventricle (RV) dysfunction,3,4 in which collagen I is dominant.4,5
Fibroblast growth factor 21 (FGF21), a member of the fibroblast growth factor family, is an endocrine factor secreted primarily by the liver; the early FGF21 experimental and clinical studies focused mainly on metabolic diseases.6–8 Recent studies have shown that FGF21 acts beneficially in systemic circulation, including cerebral and cardiac vessels,9,10 and these findings indicate that FGF21 is a potential endogenous protective factor of blood vessels. Our latest in vitro study of pulmonary circulation showed that FGF21 attenuated hypoxia-induced dysfunction and apoptosis in human pulmonary arterial endothelial cells (HPAECs). 11 However, whether FGF21 has similar beneficial effects in hypoxia-induced pulmonary hypertension (HPH) animal model remains unknown.
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated nuclear transcription factors that are classified into three subtypes: α, β/δ, and γ. 12 Among these, PPARγ has multiple pharmacological activities in the pulmonary vasculature, for example, PPARγ has anti-inflammatory effects,13,14 inhibits smooth muscle cell proliferation, 15 and alleviates endothelium dysfunction. 13 Several in vivo studies of PH revealed that PPARγ activation contributed to relieving PH by inhibiting inflammation 14 and alleviating pulmonary arterial remodeling14,16,17 and pulmonary arterial collagen deposition.14,17 These findings demonstrate that PPARγ is a protective factor against PH.
Recent studies have reported the interaction between FGF21 and PPARγ in extrapulmonary tissues.18,19 However, whether the interaction between FGF21 and PPARγ also exists in the lungs remains unknown. The present study aims to determine whether a similar role exists in HPH models.
Adenosine monophosphate-activated protein kinase (AMPK), a highly conserved serine/threonine protein kinase, is activated by phosphorylation of the α-subunit (Thr172). 20 PPARγ coactivator-1α (PGC-1α) has been reported as a key regulator of hypoxia-induced endothelial dysfunction. 21 It has been reported that FGF21 regulates energy metabolism through the AMPK/PGC-1α pathway in adipose tissues. 22 Furthermore, our previous studies proved that AMPK activation compensatively ameliorated pulmonary circulation changes triggered by chronic hypoxia, and this process played an important role in inhibiting HPH.23–25 The single-transmembrane protein β-klotho (KLB), a co-factor of FGF21, is essential for FGF21 and FGF receptor (FGFR) binding. As previously reported, KLB was up-regulated in adipose tissues by PPARγ agonists, while PPARγ siRNA decreased KLB mRNA levels.26,27 These data indicate that PPARγ is required for KLB expression. Thus, we speculated that in HPH models, FGF21 promotes PPARγ expression via the AMPK/PGC-1α pathway and that KLB may act as a key protein in PPARγ-induced FGF21 expression.
Here, we aimed to investigate whether FGF21 exerted protective effects against HPH in vivo. Moreover, we investigated the relationship between FGF21 and PPARγ in HPH models and the underlying mechanism using in vivo and in vitro studies.
Methods
Reagents
FGF21 was obtained from Peprotech (Rock Hill, NJ, USA). The PPARγ agonist rosiglitazone and PPARγ antagonist GW9662 were obtained from Selleck (Houston, TX, USA). The AMPK antagonist Compound C was obtained from Sigma (St. Louis, MO, USA). Dulbecco’s modified Eagle medium (DMEM, high glucose), streptomycin, penicillin G, and fetal bovine serums (FBS) were obtained from Gibco BRL (Gaithersburg, MD, USA). Rabbit antibodies against FGF21 (lot no. ab171941), PPARγ (lot no. ab45036 and lot no. ab209350), PGC-1α (lot no. ab54481) and collagen I (lot no. ab34710) and a mouse antibody against smooth muscle myosin heavy chain 11 (MYH11) (lot no. ab53219) were purchased from Abcam (Cambridge, UK). Rabbit antibodies against phospho-AMPK (Thr172, lot no. #2535), AMPK (lot no. #5831) and GAPDH (lot no. #5174) were purchased from Cell Signaling Technology (Beverly, MA, USA). A rabbit antibody against KLB (lot no. SAB2108630) was purchased from Sigma (St. Louis, MO, USA). A horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG antibody (lot no. BL003A) was obtained from Biosharp (Hefei, CHN). Donkey anti-rabbit IgG H&L (Alexa Fluor 594) (lot no. ab150076) and donkey anti-mouse IgG H&L (Alexa Fluor 488) (lot no. ab150105) antibodies were obtained from Abcam (Cambridge, UK). SuperSignal (R) West Femto Maximum Sensitivity Substrate, RIPA buffer, protease and phosphatase inhibitor mini tablets, and a bicinchoninic acid (BCA) protein assay kit were purchased from Pierce (Madison, WI, USA).
Animal models
Male C57Bl/6 mice (8–12 w, 20–25 g) were obtained from Vital River Laboratory Animal Technology (Beijing, CHN). The mice were given free access to food and water and housed in a specific pathogen-free (SPF) animal facility with a 12:12-h light-dark cycle and a temperature of 20–24°C and 55–65% humidity. The animal housing and experimental protocols were approved by the Animal Ethics Committee of Wenzhou Medical University. Sixty mice were randomly assigned to five groups (12 mice per group): normoxia group (N, saline-treated), hypoxia group (H, saline-treated), hypoxia plus FGF21 group (HF, FGF21, 0.4 mg/kg i.p.), hypoxia plus PPARγ agonist rosiglitazone group (HR, rosiglitazone, 10 mg/kg i.p.), and hypoxia plus PPARγ antagonist GW9662 group (HG, GW9662, 2 mg/kg i.p.). All hypoxia groups were housed in a closed hypoxia chamber (8 h per day, six days per week), and the normoxia group was exposed to room air. Intraperitoneal injections were given half an hour before each time the mice were placed into the chambers. The hypoxic environment of the chamber was established by filling it with nitrogen. The detector monitored the oxygen concentration dynamically and stabilized the concentration at 9–11% automatically. The hypoxia exposure lasted for four weeks.
Cell culture and treatment
Rat PASMCs were cultured in DMEM supplemented with 100 μg/mL streptomycin, 100 IU/mL penicillin and 10% FBS. After reaching 80–90% confluence, cells were treated with 0.25% trypsin-EDTA for further passaging. PASMCs were used at passages 4–6. The cultured cells were confirmed to be PASMCs by immunofluorescence. For further in vitro study, PASMCs were divided into the following groups: normoxia group (N), hypoxia group (H), hypoxia plus FGF21 group (HF, FGF21, 25 ng/mL), hypoxia plus FGF21 and Compound C group (HFC, Compound C, 40 μmol/L), hypoxia plus FGF21, siRNA-AMPK group (HFSA, siRNA-AMPK, 50 nmol/L), hypoxia plus rosiglitazone group (HR, rosiglitazone, 10 μmol/L), hypoxia plus rosiglitazone, siRNA-KLB group (HRSK, siRNA-KLB, 50 nmol/L). The normoxia group was cultured in a normal incubator (37°C, 21% O2 and 5% CO2/balance N2) for 24 h, while the hypoxia groups were kept in a hypoxia incubator (37°C, 5% O2 and 5% CO2/balance N2) for 24 h.
siRNA transfection
The siRNAs were synthesized by Ribobio (Guangzhou, CHN). Cells were transfected with siRNAs according to the manufacturer's protocol. The cells were incubated with the transfection complex, and the efficiency of gene knockdown was assessed after 24 h. The most efficient of the three synthesized siRNAs was selected by Western blot assay.
Invasive haemodynamic measurements
The mice were anaesthetized with 20% urethane (1 mL/100 g, i.p.) after hypoxia exposure. Two home-made polyethylene (PE) catheters (inner diameter: 0.5 mm, outer diameter: 0.9 mm) that were prefilled with heparin were inserted into the RV and left carotid artery.28,29 The right ventricular systolic pressure (RVSP) and the mean carotid arterial pressure (mCAP) were recorded by pressure transducers (PowerLab 8/35 multi-channel biological signal recording system, AD Instruments, AUS).
Right ventricular hypertrophy measurement
The mice were sacrificed by exsanguination for further examination. The hearts were dissected out and then cut along the edge of the RV and the interventricular septum with a dissecting microscope (Nikon, Tokyo, Japan) to divide hearts into two parts: the RV and the left ventricle plus septum (LV+S). The ratio of the RV to (LV+S) weights was calculated as an index to assess right ventricular hypertrophy.
Measurement of pulmonary arterial remodeling and collagen deposition
The upper lobes of the right lung were removed, fixed in 4% paraformaldehyde for 24 h, de-hydrated in a graded ethanol series, embedded in paraffin and sectioned in 5-μm-thick slices. After the sections were stained with haematoxylin-eosin (HE), the structural remodeling of the pulmonary arteries was evaluated microscopically. Pulmonary arteries with external diameters of 25–100 μm were randomly selected, and the ratios of the pulmonary artery wall area to the total area (WA/TA) and the wall thickness to the total thickness (WT/TT) were calculated and analyzed by Image-Pro Plus 6.0 (Media Cybernetics, Bethesda, MD, USA) to reflect pulmonary arterial remodeling.28–30 Additionally, the paraffin sections were Masson stained to assess the degree of collagen deposition around the pulmonary arteries. The stained sections were then observed microscopically; the collagen fibres were stained blue, and the muscle fibres, cytoplasm, and erythrocytes were red.
Immunofluorescence detection
For the immunofluorescence assays, FGF21, PPARγ, and collagen I were detected as proteins of interest, and MYH11 (a marker of PASMCs) was detected as a localization marker. After de-hydration with a sucrose gradient, the lung tissue specimens were embedded in Tissue-Tek OCT compound (Sakura, Japan). Then, 10-μm frozen sections were prepared, fixed in cold methanol for 10 min, and blocked with 5% donkey serum for 1 h before incubation with the mouse antibody against MYH11 (1:200) and the rabbit antibody against FGF21 (1:150), PPARγ (1:200) or collagen I (1:200) overnight at 4°C. After rewarming to room temperature, the sections were washed three times with phosphate-buffered saline (PBS) and then incubated with a mixture of the Alexa Fluor 594 donkey anti-rabbit IgG antibody H&L (1:500) and Alexa Fluor 488 donkey anti-mouse IgG antibody H&L (1:500) in a dark room for 1 h. The immunolabeled sections were observed at 488 and 594 nm using a laser scanning confocal microscope (Olympus, Tokyo, Japan). The fluorescence intensity ratios of the FGF21, PPARγ, and collagen I proteins were analyzed with Image-Pro Plus 6.0.
Western blot detection
Lung tissues were homogenized in cold RIPA lysis buffer containing protease and phosphatase inhibitors with an automatic homogenizer (FastPrep-24 5G, MP Biomedicals, CA, USA). After ultrasonic disruption, the lysates were centrifuged (12,000 r/min, 4°C) for 30 min, and the supernatants were collected. PASMCs were lysed with cold RIPA lysis buffer containing PMSF. The lysates were centrifuged (12,000 r/min, 4°C) for 30 min, and the supernatants were collected. The protein concentrations were determined with a Pierce BCA protein assay kit. Equal amounts of protein (60 μg) were separated with 8–12% SDS-PAGE, transferred onto PVDF membranes (Millipore, MA, USA), blocked with 5% skim milk (BD, NJ, USA), and incubated with specific primary antibodies against FGF21 (1:1000), PPARγ (lot no. ab45036, 1:500, for mouse lung homogenates), PPARγ (lot no. ab209350, 1:1000, for rat PASMCs), AMPK (1:1000), phospho-AMPK (1:1000), PGC-1α (1:1000), KLB (1:1000), and GAPDH (1:1000) overnight at 4°C. After being washed three times, the membranes were incubated with the HRP-conjugated goat anti-rabbit secondary antibody (1:10,000) for 1 h at room temperature. The immunoreactive bands were visualized with an enhanced chemiluminescence (ECL) substrate reagent (Pierce, WI, USA) and then analyzed with Quantity One v-4.6.2 software (Bio-Rad Laboratories, CA, USA).
Statistical analysis
Statistical analyses were performed with GraphPad Prism 6.0 (GraphPad Software, CA, USA). All data are expressed as the mean ± standard error of mean (SEM). Comparisons between two groups were analyzed by Student’s t-test, and multiple comparisons were analyzed by one-way analysis of variance (ANOVA). P values of <0.05 were considered statistically significant.
Results
FGF21 attenuated HPH and RV hypertrophy
To determine the effects of FGF21 on HPH, mice were exposed to hypoxia for four weeks and treated with FGF21, the PPARγ agonist rosiglitazone or the PPARγ antagonist GW9662. RVSP was recorded to reflect the PAP, and RV/(LV+S) was calculated to assess RV hypertrophy. As shown in Figure 1(a) and (c), hypoxia increased RVSP, which demonstrated that the hypoxia exposure was effective. However, this increase was inhibited by exogenous FGF21 or rosiglitazone treatment and was strengthened by GW9662 treatment. Moreover, there were no significant differences in mCAP among the five groups (Figure 1(b) and (d)). As shown in Figure 1(e), RV/(LV+S) was higher in the H group than in the N group, indicating that hypoxia exposure successfully induced RV hypertrophy. FGF21 or rosiglitazone administration significantly reduced this increase in PH mice, while GW9662 administration had the opposite effect.
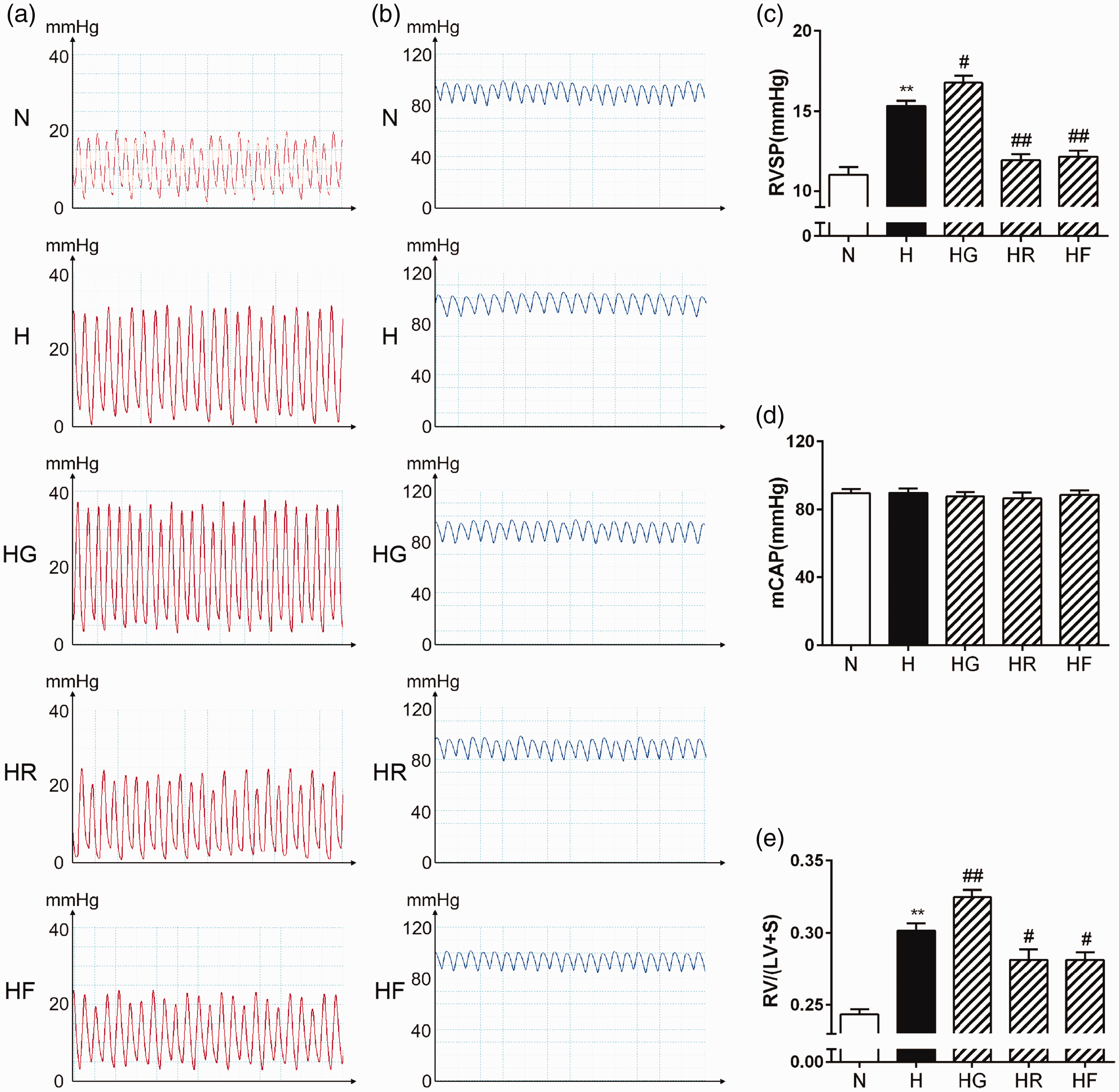
FGF21 and PPARγ attenuated HPH and RV hypertrophy. Representative images of RVSP waves (a) and mCAP waves (b) were obtained via an invasive catheterization procedure. RVSP (c; n = 11–12), mCAP (d; n = 10–12), and [RV/(LV+S)] (e; n = 10) values for each group were analyzed. The data are presented as the mean ± standard error of mean (SEM). *P < 0.05, **P < 0.01 versus the normoxia group, #P < 0.05, ##P < 0.01 versus the hypoxia group. (A color version of this figure is available in the online journal.)
FGF21 alleviated hypoxia-induced pulmonary arterial remodeling
Sections stained with HE showed that pulmonary artery wall thickening and pulmonary artery muscularization were more obvious in the H group than in the N group. These changes were alleviated after treatment with FGF21 or rosiglitazone and exacerbated after treatment with GW9662. To evaluate the role of FGF21 in pulmonary arterial remodeling, we examined the pulmonary artery (external diameters: 25–100 μm) WA/TA (%) and WT/TT (%). As shown in Figure 2, the pulmonary artery WA/TA (%) and WT/TT (%) ratios were significantly higher in the H group than in the N group. These increases were reversed by exogenous FGF21 or rosiglitazone treatment and strengthened by GW9662 treatment.
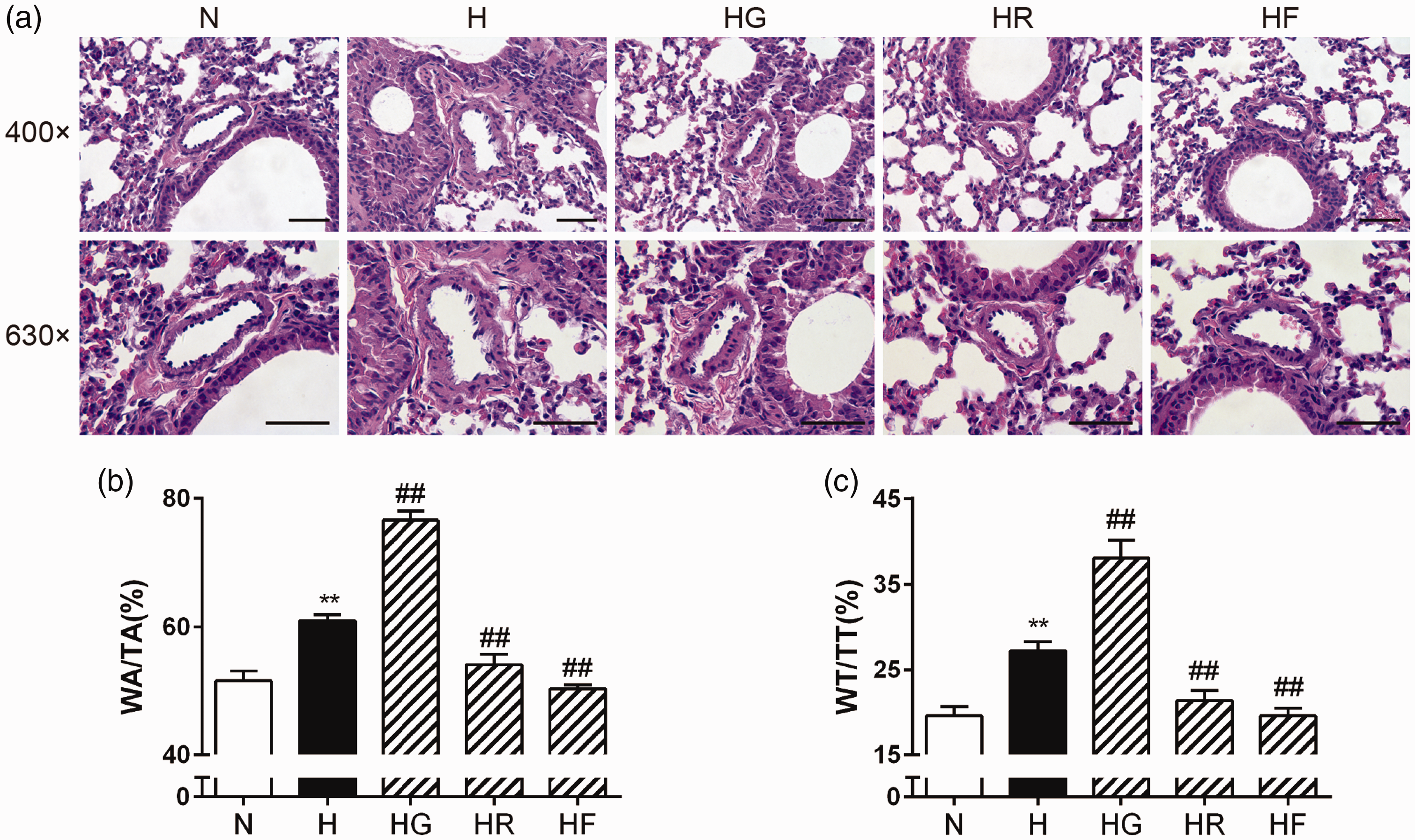
FGF21 and PPARγ alleviated hypoxia-induced pulmonary arterial remodeling and morphological changes. Lung tissue sections were stained with HE, and the morphological structures of the pulmonary arteries in each group are shown (a; scale bars indicate 50 μm). (WA/TA) (b; n = 6) and (WT/TT) (c; n = 6) values for each group were analyzed. The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01 versus the normoxia group, #P < 0.05, ##P < 0.01 versus the hypoxia group. (A color version of this figure is available in the online journal.)
FGF21 alleviated hypoxia-induced pulmonary collagen deposition
To investigate the role of FGF21 in pulmonary arterial collagen deposition, we used Masson staining to determine the collagen composition around the pulmonary arteries in each group. As shown in Figure 3(a), the amount of collagen around the pulmonary arteries was increased in the H group. A significant amount of collagen was stained in the HG group. In contrast, FGF21 or rosiglitazone administration had the positive effect of alleviating the collagen deposition around the pulmonary arteries in HPH mice.
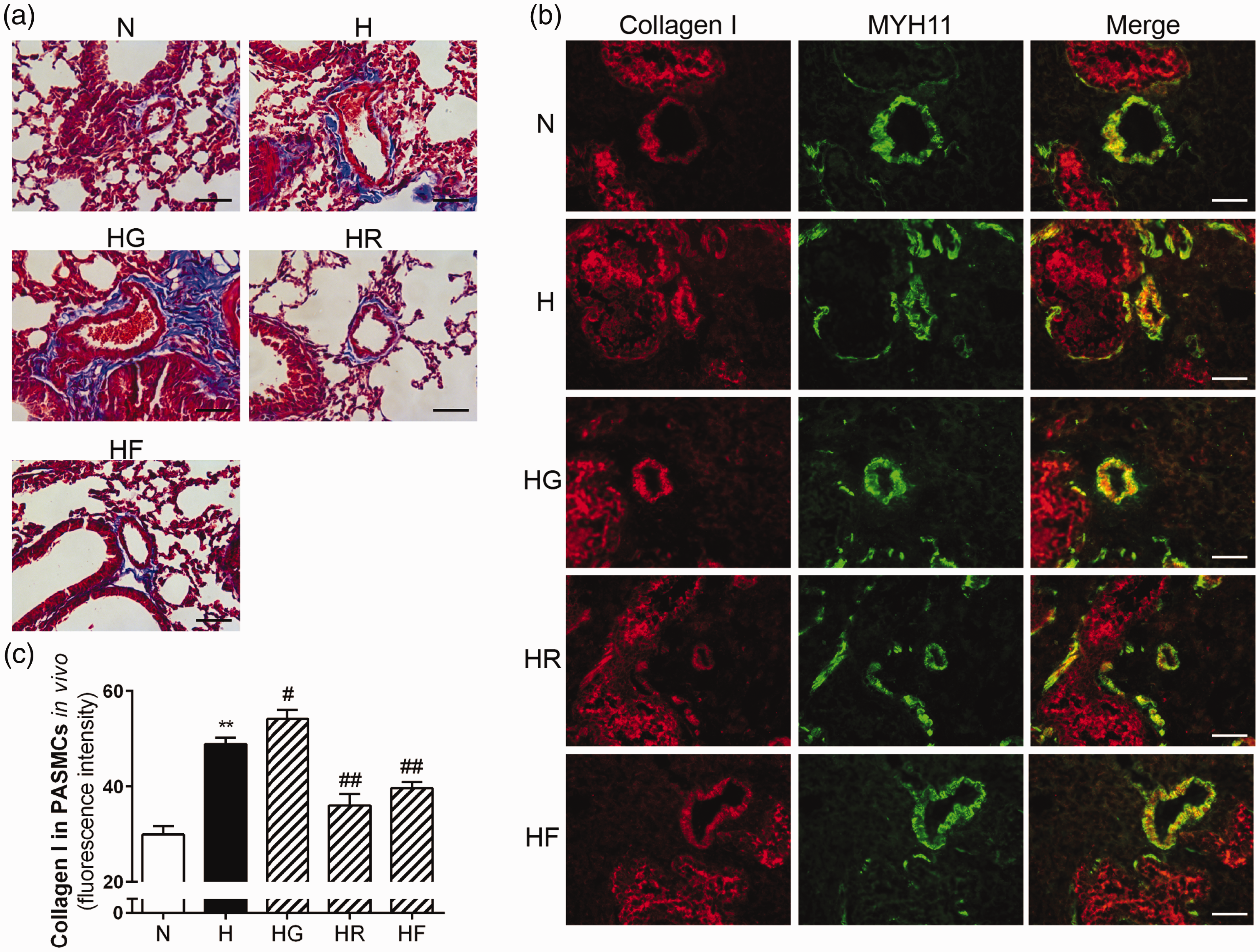
FGF21 and PPARγ alleviated hypoxia-induced collagen deposition and inhibited collagen I expression in HPH mouse lungs. The degree of collagen deposition (blue) in each group was evaluated microscopically by Masson staining (a; scale bars indicate 50 μm). Collagen I and MYH11 expression levels in mouse PASMCs for each group were assessed by immunofluorescence staining (b; 400× scale bars indicate 50 μm). Collagen I protein was stained red, and MYH11 was stained green. Collagen I protein fluorescence intensity values were calculated (c; n = 5). The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01 versus the normoxia group, #P < 0.05, ##P < 0.01 versus the hypoxia group. (A color version of this figure is available in the online journal.)
FGF21 attenuated collagen I expression in HPH mice
Collagen I, a representative type of collagen in pulmonary arteries, was chosen as an indicator to reflect the biological effects of FGF21 in ameliorating the collagen deposition in HPH. As shown in Figure 3(b) and (c), hypoxia markedly increased collagen I expression in the mouse pulmonary arteries. However, the collagen I fluorescence intensity ratio was significantly decreased after treatment with FGF21 or rosiglitazone. In contrast, collagen I expression was further enhanced in the HG group.
PPARγ promoted FGF21 expression, and FGF21 promoted PPARγ expression in HPH
To investigate the relationship between FGF21 and PPARγ in HPH, we measured FGF21 and PPARγ expression by immunofluorescence and Western blotting. As shown in Figures 4(a) and (b) and Figure 5(a) and (b), the expression levels of FGF21 and PPARγ in the pulmonary arteries were lower in the H group than in the N group. These decreases were reversed by exogenous FGF21 or rosiglitazone treatment. In contrast, the FGF21 and PPARγ fluorescence intensity ratios were even lower in the HG group than in the H group. The Western blots of the total lung homogenates indicated the same results (Figures 4(c) and 5(c)). These findings demonstrated that exogenous FGF21 administration (i.p.) increased FGF21 expression in the pulmonary arteries and lung homogenates of HPH mice. Rosiglitazone or GW9662 treatment effectively activated or inhibited PPARγ in HPH mice. PPARγ activation increased FGF21 expression, while PPARγ inhibition reduced FGF21 expression in HPH mice. Moreover, exogenous FGF21 administration also increased PPARγ expression in HPH mice. These data confirm the mutual promotion of FGF21 and PPARγ in HPH mouse lungs.
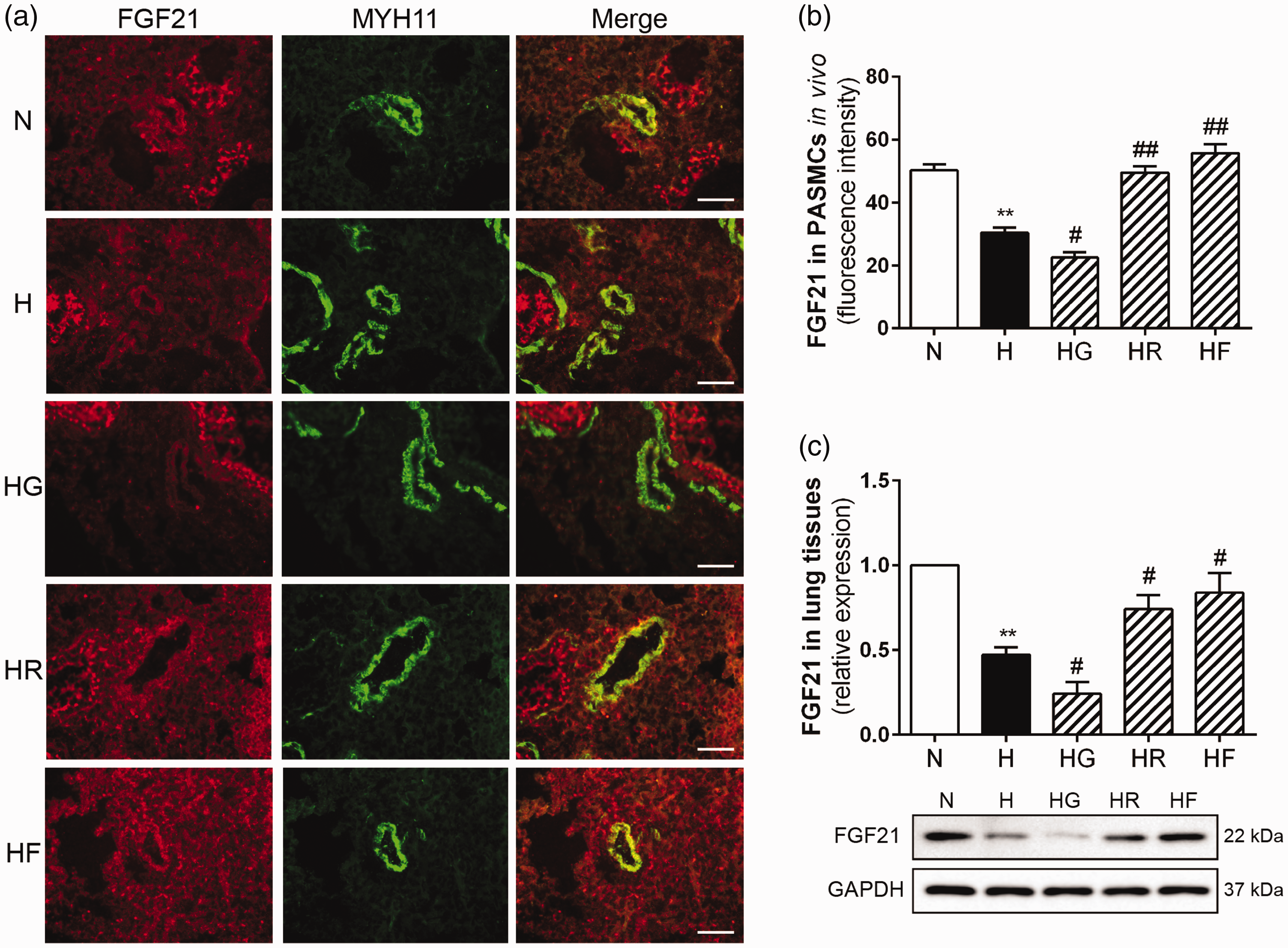
PPARγ promoted FGF21 expression in HPH mouse lungs. FGF21 and MYH11 expression levels in mouse PASMCs for each group were assessed by immunofluorescence staining (a; 400× scale bars indicate 50 μm). FGF21 protein fluorescence intensity values were calculated (b; n = 5). FGF21 protein expression levels in lung homogenates were examined by Western blotting, and GAPDH was used as an internal control (c; n = 3). The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01 versus the normoxia group, #P < 0.05, ##P < 0.01 versus the hypoxia group. (A color version of this figure is available in the online journal.)
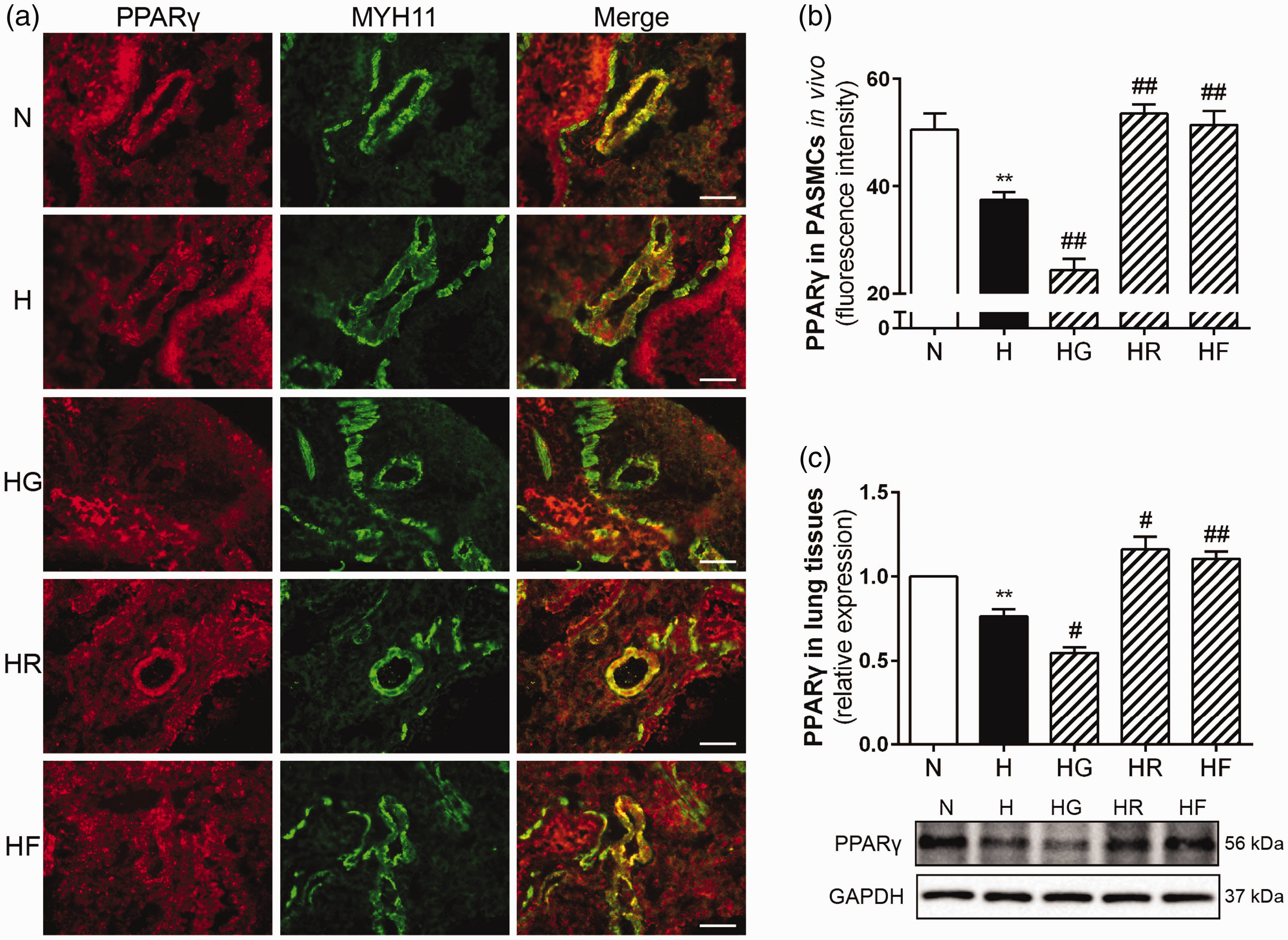
FGF21 promoted PPARγ expression in HPH mouse lungs. PPARγ and MYH11 expression levels in mouse PASMCs for each group were assessed by immunofluorescence staining (a; 400× scale bars indicate 50 μm). PPARγ protein fluorescence intensity values were calculated (b; n = 5). PPARγ protein expression levels in lung homogenates were examined by Western blotting, and GAPDH was used as an internal control (c; n = 3). The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01 versus the normoxia group, #P < 0.05, ##P < 0.01 versus the hypoxia group. (A color version of this figure is available in the online journal.)
FGF21 promoted PPARγ expression via the AMPK/PGC-1α pathway
To investigate how FGF21 promoted PPARγ expression in HPH, Western blot analyses of protein in HPH mouse lung homogenates and hypoxic rat PASMCs were performed. As shown in Figure 6(a), hypoxia increased AMPK phosphorylation in mouse lung homogenates, and exogenous FGF21 administration further enhanced this increase. Our results also showed that the PGC-1α protein level was decreased by hypoxia, and FGF21 significantly reversed this decrease in HPH mice (Figure 6(b)). In vitro, as shown in Figure 6(c), (d), (f) and (g), hypoxia reduced PPARγ and PGC-1α protein expression in rat PASMCs, while FGF21 treatment reversed these decreases. Importantly, when hypoxic PASMCs were treated with Compound C or siRNA-AMPK, the FGF21-induced increases of PPARγ and PGC-1α protein expression were inhibited.
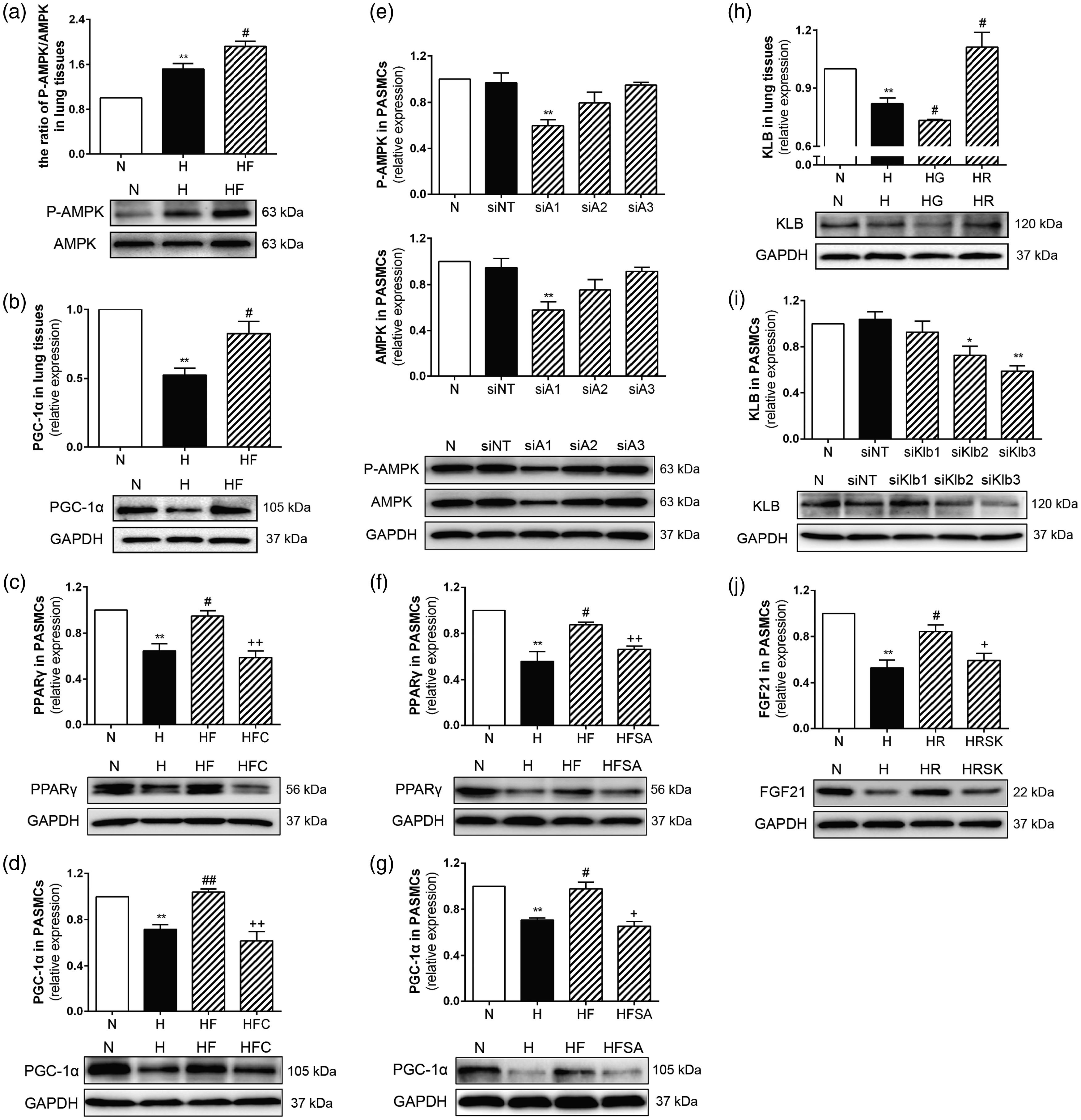
FGF21 promoted PPARγ expression via the AMPK/PGC-1α pathway, and PPARγ promoted FGF21 through KLB protein induction in HPH. Western blotting revealed the protein expression of phospho-AMPK, AMPK, PGC-1α, KLB, FGF21, PPARγ in lung homogenates and/or PASMCs. GAPDH was used as an internal control. FGF21 increased AMPK phosphorylation and PGC-1α expression in PAH mice lung homogenates (a, b; n = 3). The siRNA showing the highest efficiency of AMPK knockdown was selected by Western blot assay (e; n = 3). FGF21 increased PPARγ and PGC-1α expression in hypoxic PASMCs, and Compound C or siRNA-AMPK reversed the up-regulation (c, d, f, g; n = 3). Rosiglitazone increased KLB expression, and GW9662 decreased KLB expression in PAH mice lung homogenates (h; n = 3). The siRNA showing the highest efficiency of KLB knockdown was selected by Western blot assay (i; n = 3). Rosiglitazone increased FGF21 expression in hypoxic PASMCs, and siRNA-KLB reversed this up-regulation (j; n = 3). The data are presented as the mean ± SEM. *P < 0.05, **P < 0.01 versus the normoxia group, #P < 0.05, ##P < 0.01 versus the hypoxia group, +P < 0.05, ++P < 0.01 versus the hypoxia plus FGF21 group or the hypoxia plus rosiglitazone group.
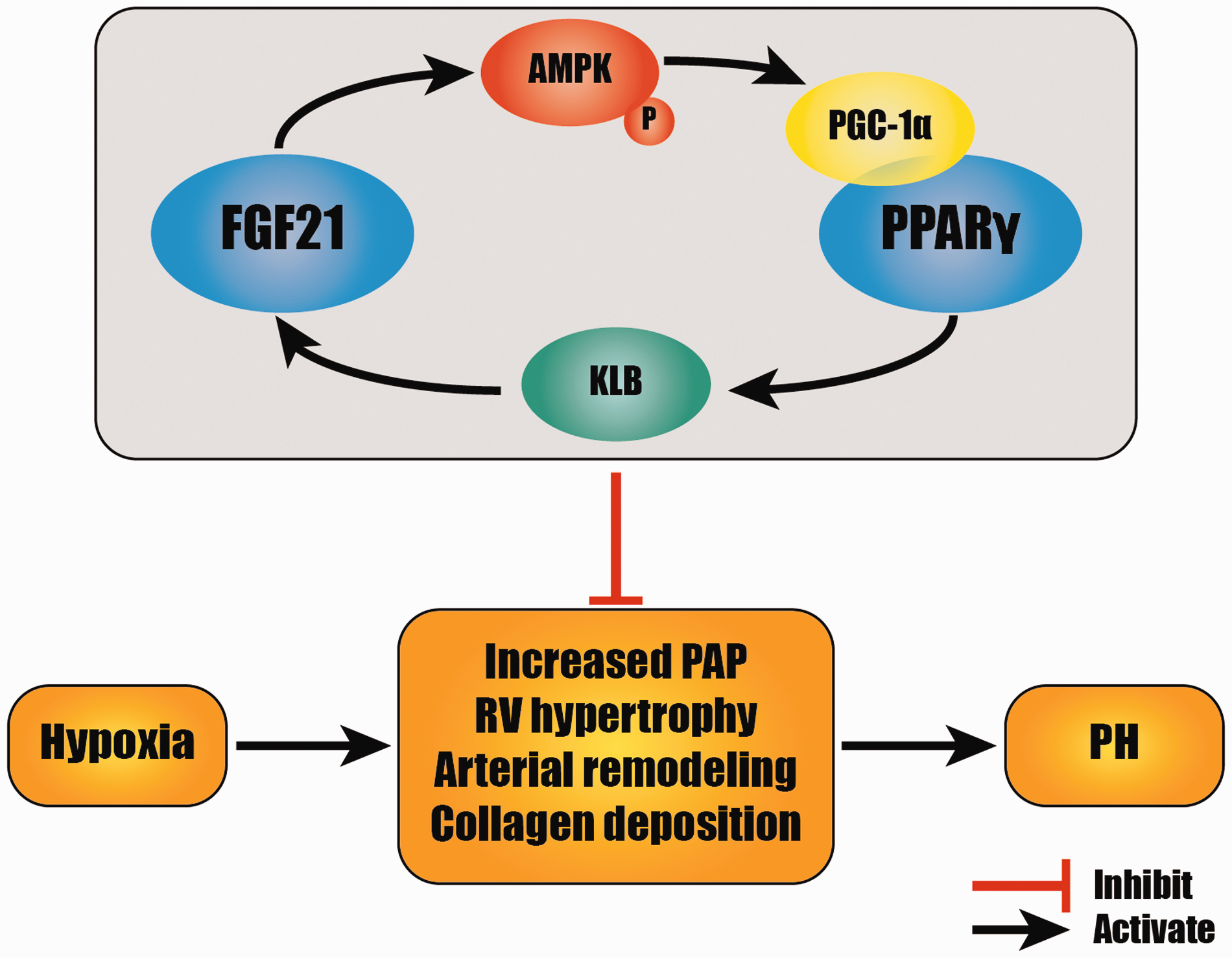
FGF21 effectively attenuated HPH by inhibiting increased PAP, RV hypertrophy, pulmonary arterial remodeling, and collagen deposition. The mutual promotion of FGF21 and PPARγ in which the AMPK/PGC-1α pathway and KLB protein are involved may play an important role in these protective effects. (A color version of this figure is available in the online journal.)
PPARγ promoted FGF21 expression through KLB protein
To explore how PPARγ promoted FGF21 expression in HPH, Western blot analyses of protein in HPH mouse lung homogenates and hypoxic rat PASMCs were performed. As shown in Figure 6(h), hypoxia significantly decreased KLB protein expression in mouse lung homogenates. Rosiglitazone treatment increased the KLB protein levels in the HPH mouse lung homogenates, while GW9662 treatment further decreased the KLB protein expression levels in the HPH mouse lung homogenates. In vitro, the siRNA showing the highest efficiency of KLB knockdown was selected by Western blot assay. The selected siRNA-KLB clearly and significantly down-regulated the KLB protein expression in rat PASMCs (Figure 6(i)). As shown in Figure 6(j), hypoxia inhibited the FGF21 protein expression in rat PASMCs, and rosiglitazone reversed this decrease. It was noteworthy that the rosiglitazone-induced increase of FGF21 expression in hypoxic PASMCs was inhibited by siRNA knockdown of KLB.
Discussion
The HPH animal model has been widely used in experimental studies. This model is characterized by increased PAP, pulmonary arterial remodeling, and right ventricular hypertrophy. In the present study, we exposed mice to hypoxia for four weeks (8 h per day, six days per week) as previously reported.5,29 Increased RVSP, RV/(LV+S), WA/TA (%) and WT/TT (%), as well as histopathological changes, demonstrated that the HPH model was established successfully.
PH is a progressive and serious pathological phenomenon with a poor prognosis, and current therapies are highly limited. Current PH treatments remain expensive and are more palliative than curative; these treatments yield few long-term benefits and barely reduce mortality. 31 Thus, the pressing need for more effective and less expensive therapies is critical. Recent studies have highlighted the therapeutic value of FGF21 in the vascular protection of the systemic circulation.9,10 Because of the similarity between the pulmonary circulation and the systemic circulation, we previously performed an in vitro experiment to explore the role of FGF21 in the pulmonary circulation. Consistent with our hypothesis, our previous study confirmed the protective effect of exogenous FGF21 in hypoxia-treated HPAECs. 11 Therefore, FGF21 is hypothesized to act as a novel vascular protective factor for treating PH. Our present study found that exogenous FGF21 treatment attenuated hypoxia-induced increases in RVSP and RV/(LV+S) and ameliorated pulmonary arteriolar thickening and luminal stenosis. We also found that hypoxia-induced pulmonary arterial collagen deposition was attenuated by exogenous FGF21, and this finding was consistent with a mesangial cell study. 32 Herein, we reported for the first time that FGF21 alleviated HPH by attenuating the increased PAP, pulmonary arterial remodeling, and collagen deposition in vivo.
The role of PPARγ in PH has been widely explored. Several in vivo studies have confirmed that PPARγ agonists effectively alleviate PAP, pulmonary arterial remodeling, pulmonary arterial collagen deposition, RV hypertrophy, and dysfunction in monocrotaline-induced PH and/or HPH.14,16,17,33 So far, PPARγ agonists have not been used clinically for treating PH. In the present study, we confirmed the protective effects of PPARγ in HPH mice by up-regulating and down-regulating PPARγ. Our data provide evidence for the clinical application of PPARγ agonists for treating PH.
Moyers et al. 18 reported that treating adipocytes with FGF21 for 72 h induced an increase in PPARγ protein expression; moreover, rosiglitazone, a PPARγ agonist, enhanced FGF21 activity. Muise et al. 34 identified 33 genes that were robustly regulated by PPARγ agonists that code for secreted proteins, one of which was FGF21. These results reveal the interaction between FGF21 and PPARγ. However, evidence of this interaction in the lungs is missing. In the present study, we found that hypoxia decreased FGF21 and PPARγ expression in HPH mice. Exogenous FGF21 treatment reversed this decrease in PPARγ a PPARγ agonist reversed the decrease in FGF21, while a PPARγ antagonist further reduced FGF21 expression. For the first time, our results confirm this mutual promotion of FGF21 and PPARγ in HPH, and our results are consistent with those reported in extrapulmonary studies. Our findings provide evidence for the combination of FGF21 and PPARγ agonists to treat PH, and this combination could reduce dosage and side effects.
Recent studies have revealed that FGF21 plays a role in adipose tissues and the testis through the AMPK/PGC-1α pathway.22,35 Here, we used Western blot assays to show that FGF21 obviously enhanced hypoxia-induced AMPK phosphorylation and reversed the hypoxia-induced decrease in PGC-1α protein levels in mouse lung homogenates. More importantly, we found that in hypoxic PASMCs, FGF21 failed to promote PPARγ and PGC-1α expression when AMPK was blocked or down-regulated. AMPK activation has been demonstrated to increase PGC-1α downstream. However, we found it interesting that hypoxia resulted in an increase in AMPK phosphorylation with a decrease in PGC-1α expression, which was consistent with the previous study 21 in PAECs. Ramjiawan et al. 36 reported the biphasic PGC-1α expression in response to hypoxia. They found that PGC-1α down-regulation during hypoxia was due to histone deacetylation, and longer hypoxia increased AMPK phosphorylation which reversed this decrease of PGC-1α. We surmised that hypoxia affected PGC-1α not only via AMPK, but through other pathway, histone deacetylation for example, and the effects of AMPK activation were not dominating in our study. PPARγ has been reported to promote the transcription and translation of KLB.26,27 We found that KLB expression in HPH mice lung homogenates was increased by rosiglitazone and further reduced by GW9662. In hypoxic PASMCs, rosiglitazone failed to increase FGF21 protein expression upon KLB knockdown. These findings reveal the underlying mechanism of the mutual promotion of FGF21 and PPARγ in HPH. FGF21 promotes PPARγ expression via the AMPK/PGC-1α pathway in HPH, and KLB acts as a key protein in the PPARγ-mediated promotion of FGF21 expression.
The present study demonstrated the beneficial effects of FGF21 in alleviating HPH. Further studies with descending dosages of FGF21 have been taken into consideration. Moreover, the present study confirms the mutual promotion of the two protective factors FGF21 and PPARγ in HPH. An efficacy study of the combination of FGF21 and a PPARγ agonist has been included in our next experimental protocol. This study could provide evidence for decreasing the drug doses. In addition, the present study found that the AMPK/PGC-1α pathway and KLB protein participate in the mutual promotion of FGF21 and PPARγ in HPH.
In summary, as shown in Figure 7, FGF21 is effective in inhibiting HPH through attenuating increased PAP, pulmonary arterial remodeling, and collagen deposition. These results provide novel insight into potential clinical therapies for PH. FGF21 and PPARγ mutually promote each other’s expression in HPH via the AMPK/PGC-1α pathway and KLB protein. These findings thus establish the possibility of using this drug combination and potential dosage reductions in clinical settings.
Footnotes
Authors’ contributions
GXC, XYH and LXW designed and performed the experiments, analyzed the data and wrote the paper; MXL and KTH participated in the study design and helped draft the manuscript; JJL, MSC and XCL performed the experiments and collected the data; MBW and LHS collected the data and performed the analyses. All authors read and approved the final manuscript.
DECLARATION OF CONFLICTING INTERESTS
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
FUNDING
This study was supported by grants from the Natural Science Foundation of Zhejiang Province, China (Y17H010028), the Project of Health Department of Zhejiang Province, China (2016DTA005), and the Chinese National Natural Science Foundation (81873411).