
Abstract
Bax is an essential mediator of mitochondria-dependent programed cell death. Bax belongs to the Bcl-2 family of proteins and its activities are regulated through interaction with other member proteins in the Bcl-2 family. To date, several apoptosis-inducing drugs activating Bax have been developed, and some of them are already in the market as therapeutics against cancer. However, at present, there are no clinically effective pharmacological Bax inhibitors protecting essential cells. Previously, we developed Bax-Inhibiting Peptides (BIPs) that belong to the peptide group of Cell-Penetrating Peptides (CPPs). CPPs have the ability to deliver cargo molecules into the cell. In this review, we will describe the mechanism of action of BIPs together with the recent applications of BIPs in disease models in vitro and in vivo. However, BIPs have several limitations in their use to treat human diseases, and other types of Bax inhibitors need to be developed for future therapeutics. Recently, several groups reported the successful development of novel small compounds inhibiting Bax. We will review these Bax inhibitors to discuss current strategies to develop pharmacological Bax inhibitors.
Impact statement
Bax induces mitochondria-dependent programed cell death. While cytotoxic drugs activating Bax have been developed for cancer treatment, clinically effective therapeutics suppressing Bax-induced cell death rescuing essential cells have not been developed. This mini-review will summarize previously reported Bax inhibitors including peptides, small compounds, and antibodies. We will discuss potential applications and the future direction of these Bax inhibitors.
Introduction
Bax is a pro-apoptotic protein that belongs to the evolutionarily conserved Bcl-2 family of proteins (reviewed in Korsmeyer 1 and Kale et al. 2 ). Bax is a main mediator of mitochondria-dependent programed cell death, and it induces necrotic cell death in certain cases even if caspase activation is inhibited. 3 At present, there is no clinically effective Bax inhibitor available. Previously, we developed Bax-inhibiting peptide (BIP), which rescues cells from Bax-induced cell death.4–7 In this article, we will review the mechanism of cell entry of BIP, the potential applications of BIP based on recent reports utilizing BIP in animal disease models, and the recent progress in pharmacological inhibition of Bax by small molecules.
Bax-inhibiting peptides
Ku70 is a multifunctional protein that is essential for non-homologous end joining DNA repair (reviewed in Downs and Jackson 8 ). It has also been shown to bind and inhibit Bax. 6 This action inhibits the conformation change of Bax (the N-terminus exposure associating with Bax activation) and translocation from the cytosol into the mitochondria.4,7,9,10 BIPs were derived from the Bax binding domain of Ku70.4,7,9 These BIPs were designed to be cell-penetrating, anti-apoptotic peptides and have been generated from a variety of species including human, rat and mouse4,7 (Figure 1). We designed a series of penta-peptides by scrambling the amino acid sequence of the Bax binding domain of Ku70 that were intended to serve as negative control peptides that retain cell-penetrating activities. Together with BIPs, these cell-penetrating penta-peptides are named cell-penetrating penta-peptides (CPP5s).5–7 The amino acid sequences of CPP5s are shown in Figure 1.
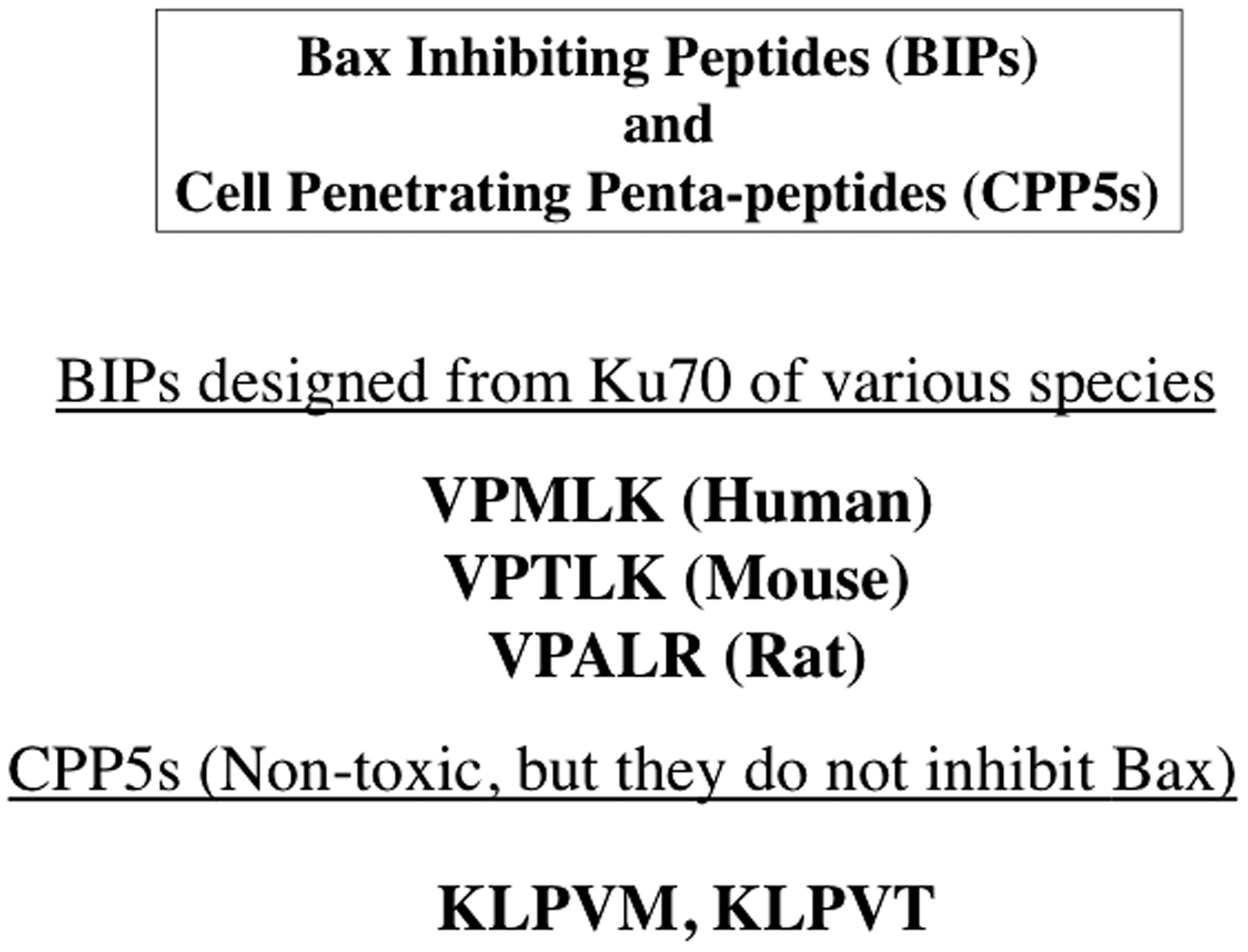
Amino acid sequences of the representative CPP5s. V: Valine; P: Proline; M: Methionine; L: Leucine; K: Lysine; R: Arginine; A: Alanine; T: Threonine.
The precise mechanism by which CPPs penetrate the plasma membrane and enter the intracellular space is not yet fully understood. The present understanding is that many CPPs use various cellular pinocytotic pathways to enter mammalian cells.7,11–14 The first generation of CPPs was developed through the discovery of the cell entry activity of the human immunodeficiency virus (HIV) TAT transactivator protein.15,16 The cell-penetrating TAT peptide (10–14 amino acids (a.a.)) was developed after this discovery.17–19 Furthermore, another type of CPP, penetratin, was developed from the homeobox domain (α-helix) of Drosophila’s antennapedia protein.20,21 Penetratin has the ability to penetrate the plasma membrane of both mammalian cells and insect cells.20,21 Since then, new CPPs other than TAT and penetratin have been discovered. These include synthetic arginine rich peptides (8 a.a.),14,22,23 the amphiphatic peptides (18 a.a.),11,24 transportan (21 a.a.),11,25,26 and others.27–29 Studies have demonstrated protein transduction activity by these peptides, which means these CPPs have the ability to carry a cargo protein (e.g. 50 kDa immunoglobulin) from culture medium into the intracellular space.27–29 While the mechanism by which CPPs penetrate cells is not yet fully understood, the sequences of amino acids contain some clues to a mechanism of penetration. The positively charged amino acids of TAT and poly-arginine might interact with either negatively charged cell surface molecules such as proteoglycan, 30 or they may interact with negatively charged phospholipids at the cell membrane surface. 28 Endocytosis or macropinocytosis of CPPs and their cargo may be triggered by these interactions. However, the passage of CPPs in the endosome through the endosome membrane into the cytosol still leaves mechanistic questions to answer.
CPP5s: Mechanism of cell entry
In terms of CPP5s (including BIPs), we previously reported that non-tagged CPP5s may require receptor-mediated cell entry. 7 This is because the dose-dependent cell entry of CPP5s (measured by Mass Spec analysis of isotope-labeled CPP5s in the cell) showed a sigmoid curve that is similar to receptor-mediated ligand cell entry such as in the internalization of a hormone by its receptor 7 (Figure 2). By this Mass Spec-based method, it was shown that the intracellular concentration of BIP becomes 6 µM when the medium concentration of the peptides becomes higher than 750 µM.7 Unlike other CPPs such as TAT and polyariginine, CPP5s did not show toxicity even at 1.6 mM in the culture medium (TAT and R8 showed toxicity at the concentration higher than 10 µM in HeLa cell and mouse embryonic fibroblasts (MEFs) culture). The remaining important problem is to identify the putative receptor mediating CPP5 cell entry. At present, such a study has not yet been accomplished. In the case of cationic CPPs such as TAT and poly-arginine peptides, cell surface proteoglycans and other negatively charged cell surface molecules are proposed to be the receptor for cell entry.28,30 These negatively charged cell surface proteins might not be the receptors for CPP5s since CPP5s are not positively charged, in contrast to cationic peptides.
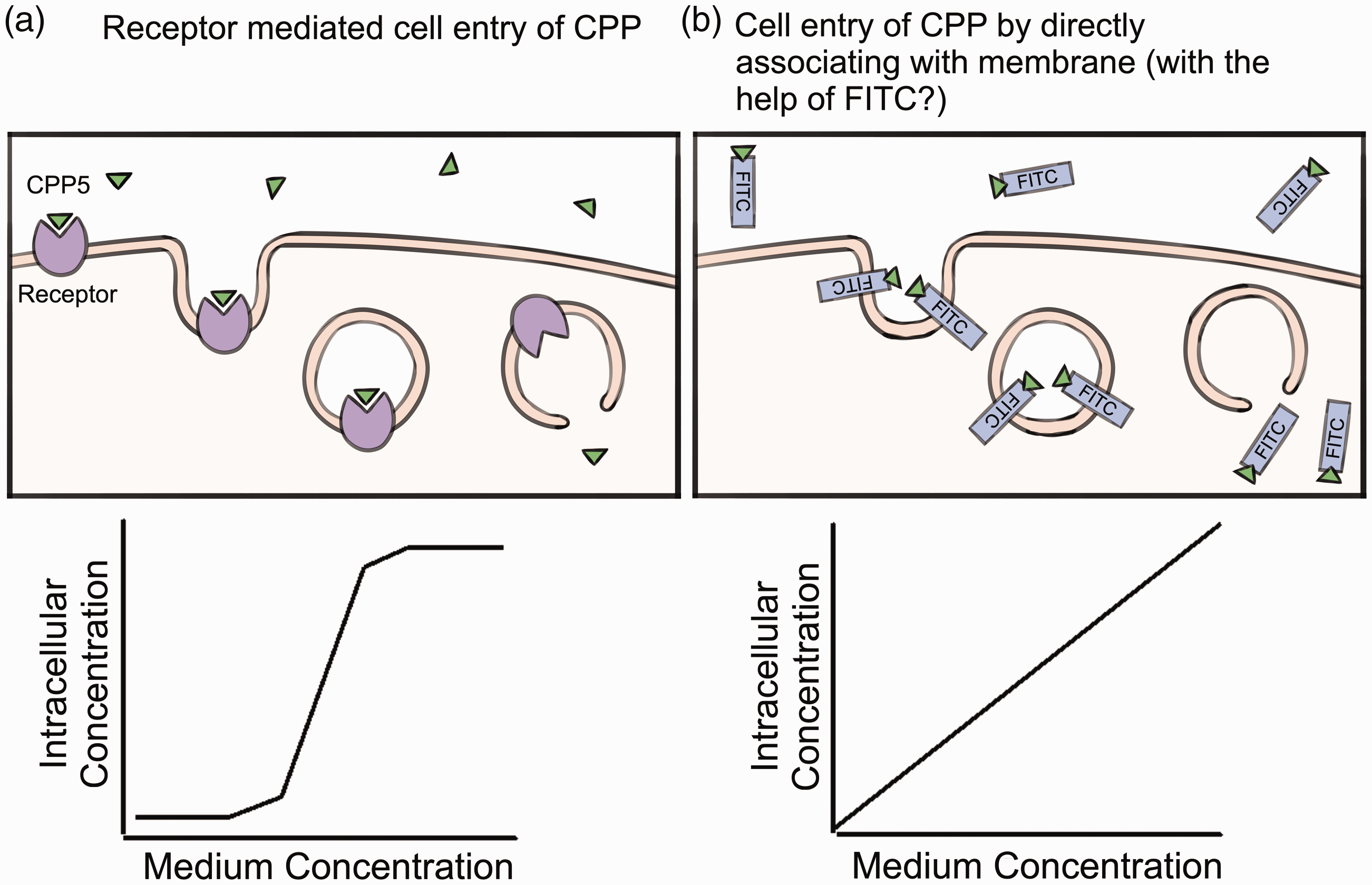
The hypothetical mechanisms of CPP5 cell entry are shown. (A color version of this figure is available in the online journal.)
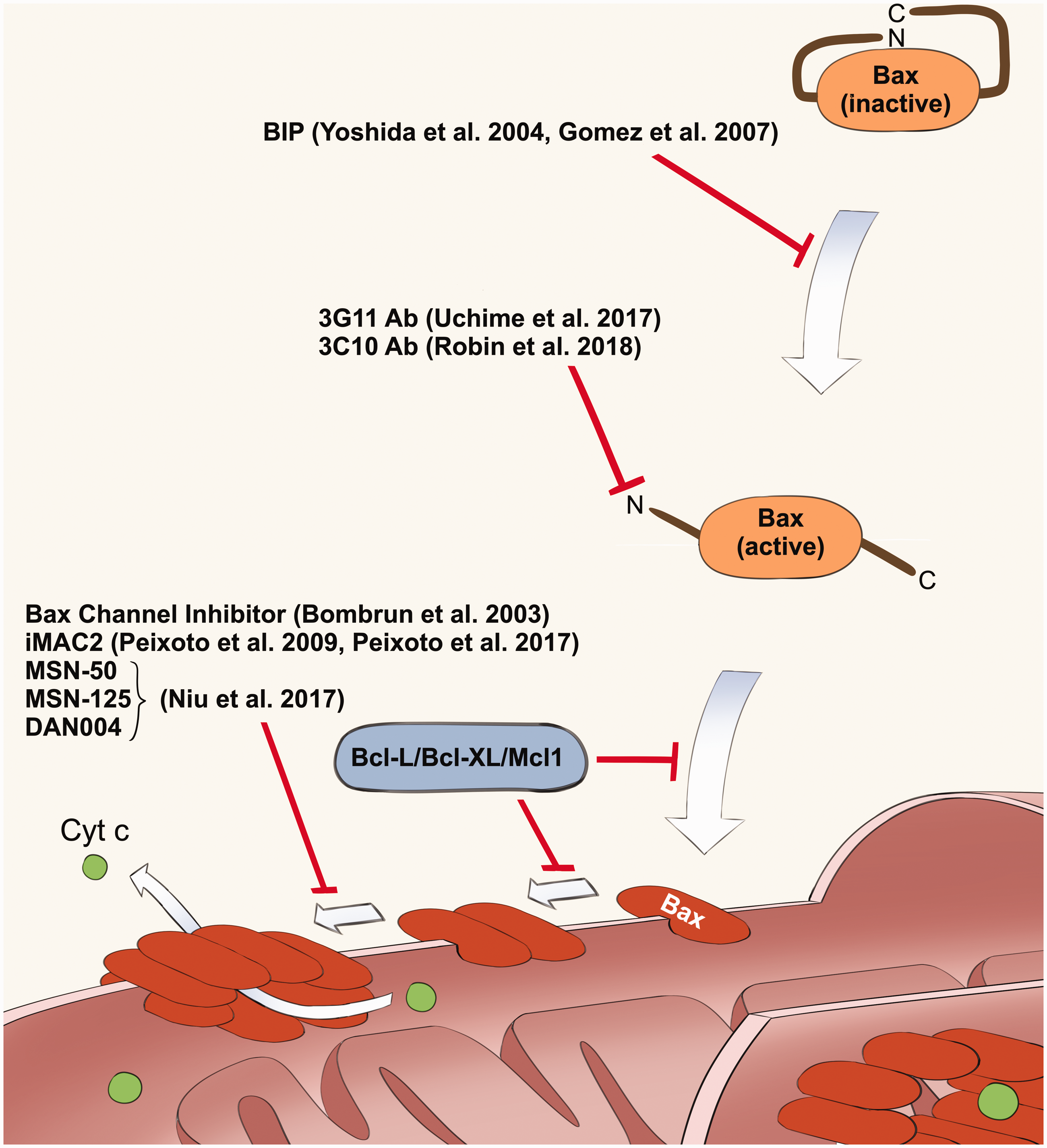
The list of Bax inhibitors explained in this article. This figure shows the multiple steps of Bax activation mechanism, and the points of regulation by these inhibitors. Anti-apoptotic members of Bcl-2 family of proteins (Bcl-2, Bcl-X, and Mcl-1) are known to inhibit mitochondrial translocation and dimerization of Bax. (A color version of this figure is available in the online journal.)1,2
On the other hand, FITC-labeled CPP5s showed a different mode of cell entry. FITC is a fluorescent chemical that is often used as a tag to trace the position of tagged molecules in the animal body as well as in cells. FITC has a hydrophobic activity, which creates an affinity to the plasma membrane and thus potentially allows FITC to accelerate the cell entry of CPPs. In the case of FITC-labeled BIPs and CPP5, the concentration of intracellular FITC-labeled BIP increased linearly when FITC-peptides were added to the culture medium up to 1.6 mM (Figure 2).7 In many cases, the FITC-tag is used to confirm the cell entry of CPPs. If the purpose is to confirm the delivery of the cargo or peptide itself, FITC-labeling is a very useful tool. However, as discussed above, it is important to note that the FITC-tag seems to enhance cell entry of CPP5s and changes the mechanism of cell entry when compared to non-tagged CPP5s.
Application of BIP for experimental models of neurological disorders
Bax knockout mice show a significant increase of the brain size and weight due to the increased survival of neurons during development.31–33 However, the mutant mouse does not develop other major abnormalities except defects in spermatogenesis and an extended female reproductive period.31,32 This phenotype suggests that Bax plays a non-replaceable role in the nervous system as an inducer of programed cell death. Interestingly, a majority of the reports of the effectiveness of BIP in disease models are related to nervous system disorders. Since Bax is the main contributor of programed cell death in the nervous system, BIP may show more prominent effects in the nervous system than in other tissues and organs. Bax deletion has been shown to have a neuroprotective effect in a variety of injury mechanisms in both in vitro and in vivo studies. 34 Bax has been shown to be upregulated in mechanisms of acute neuronal injury such as ischemic stroke and traumatic brain injury and plays a particularly important role in delayed cell death following the initial insult. 35 For example, Bax knockout mice subjected to 5 minutes of common carotid occlusion at postnatal day 7 demonstrated significantly less hippocampal neuronal loss than their wild-type counterparts. 36
BIPs have potential utility in treating a wide array of neurological disorders. BIP administration in postnatal day 9 mice that had undergone left carotid ligation decreased brain injury by 41.2% five days after the hypoxemic ischemic injury. 37 Additionally, BIP-treated mice had improved sensorimotor and motor function seven weeks after the ischemic event. While a severe ischemic injury may cause irreversible death within the affected core, administration of a Bax inhibitor near the time of injury may be able to limit the damage caused by the event by preventing apoptosis in neighboring cell populations.
In addition to rescuing cells during acute injury, BIPs may also be capable of decreasing cell death and disease progression in neurodegenerative disorders. Alzheimer’s disease is hypothesized to be caused by excess deposition of β-amyloid (Aβ) which is capable of inducing neuronal cell death in the hippocampus, though its precise mechanism is still debated. 38 In hippocampal slice cultures derived from Bax knockout mice, Aβ-induced neuronal cell death was significantly reduced compared to that of wild-type mice. 39 Additionally, administration of BIP decreased cell death in hippocampal slices treated with Aβ. 39 These results suggest that inhibition of Bax may be an effective strategy in treating Alzheimer’s disease.
Bax has also been implicated as a key player in polyglutamine (PolyQ) disorders such as Machado-Joseph disease. 9 These PolyQ mutations are believed to have toxic gain of function, which has been demonstrated to stimulate Ku70 acetylation. 9 This in turn promotes Bax activation and apoptosis. 9 BIP prevented the Bax conformational change induced by PolyQ expression. BIPs and other Bax inhibitors may therefore have potential as therapeutics for PolyQ disorders.
Application of BIP for experimental models of retinal degenerative diseases
BIPs may also be useful in the treatment of several retinal degenerative diseases. Age-related macular degeneration and Stargardt’s disease are both associated with gene alterations that result in the ineffective clearance of all-trans-retinal (atRAL).40,41 Bax activation has been shown to be an early and important step in apoptosis resulting from atRAL toxicity.42,43 The apoptosis caused by atRAL was significantly attenuated with the addition of BIP.42,43 Similarly, in an ex-vivo model derived from cultured mouse retinal tissue, BIP pre-treatment decreased cell death resulting from atRAL exposure. 43
BIP also protects retinal ganglion cells following optic nerve transection. 44 When the optic nerve of Wistar rats was transected, intravitreal injection of BIP resulted in significantly greater survival of retinal ganglion cells. This increased survival was further enhanced when the intravitreal injection of BIP was repeated on day 3 following transection. 44 This suggests that BIP may have utility in treating disorders of optic nerve injury.
Additionally, BIP protects retinal cells from hypoxic-ischemic injury, which is implicated in the development of glaucoma. 45 Hypoxia induced retinal cell death appears to occur primarily through apoptotic pathways, and rat RGCs subjected to hypoxia showed increased viability when treated with BIP. 45 All together, these studies demonstrate that BIP and the inhibition of Bax have potential as therapeutic agents in retinal disorders.
Application of BIP for experimental models of non-neurological disorders
Ischemia and perfusion (I/R)-induced cell death is the main cause of poor outcomes after the treatment of cardiac arrest and stroke. Since Bax-induced apoptotic and necrotic cell death has been implicated as a cause of major problems in the cardiovascular diseases,3,46–48 Bax inhibitors may improve results of treatment when added to the current standard of care. Recently, Suzuki et al. reported that BIP attenuated the lung fibrosis induced by bleomycin. 49 Bleomycin-induced lung fibrosis is used as a mouse model of idiopathic pulmonary fibrosis (IPF). 50 The lung fibrosis in this model is explained as the result of the local inflammatory reaction against bleomycin-induced cell death, which is caused by genotoxic stress (bleomycin is a DNA damage inducer). 50 The authors suggested that BIP may be able to block the vicious cycle of “cell death and fibrosis.” 49 In this model, the temporary protection of lung epithelial cells by BIP helped stop the local inflammatory reaction against dying epithelial cells. It thus appears that Bax inhibition can mitigate the development of IPF. In this BIP-treated mouse, Bax inhibition was temporary, so the abnormal increase of fibroblasts did not occur during the later phase. In contrast, continuous cell death inhibition (e.g. in the case of Bax knockout mice) after bleomycin-treatment may cause the problem to worsen due to increased survival of fibroblasts. For example, in one of the DNA-repair defect mutant mice (i.e. Ku70 KO mouse), Bax gene knockout resulted in the development of pulmonary hypertension-like diseases due to the abnormally increased number of endothelial cells and lung epithelial cells, as well as fibroblasts.33,51
Ex vivo application of BIP (organ transplant and cell storage)
One of the major obstacles of organ transplant is the time constraint caused by transportation from donor to recipient. Even a 1 or 2 h extension of this time limit can change the future of patients who need the matched donors’ organ in a remote location, requiring transport time that exceeds the current time limit. The types of cell death that occur after organ transportation are similar to ones that are observed in ischemia/reperfusion-induced cell death in the heart and brain after the recovery treatments of cardiac arrest and stroke, respectively. At present, Bax-induced cell death (apoptotic and necrotic cell death) and ferroptosis (a form of Caspase-independent necrotic programmed cell death are considered to be the major types of cell death in I/R-induced tissue injury.3,46–48,52 BIP and other Bax inhibitors are expected to partially protect these organs, since these inhibitors will not be able to inhibit ferroptosis in theory. Although it is not 100% protection, the increased survival in cells treated with Bax inhibitors will improve the outcomes of organ transplantation. Previously, Tanaka et al. examined the effectiveness of BIP in improving the survival of monkey kidney cell transplants. 53 In this experimental model, BIP protected the transplanted monkey kidney cells. 53 This previous study proves the principle that the pharmacological inhibition of Bax can improve the health condition of patients after I/R-induced tissue/organ injury.
In the reproductive biotechnology field, the maintenance of oocyte viability is an essential issue for in vitro fertilization, transgenic animal production, and other embryo manipulation-based biotechnology. Bax KO female mice exhibit a longer reproductive life span 31 and the Bax-deficient oocytes showed resistance to apoptotic stresses. 31 Therefore, the use of a Bax inhibitor is expected to improve the survival of oocytes cultured in vitro. In fact, BIP (100 µM) has been shown to improve the survival of bovine oocytes in vitro, and BIP was able to improve the success rate of blastocyst formation. 54 BIP may become a useful tool as a cyto-protective additive to preserve cells and tissues ex vivo.
The potential application of CPP5 and BIP as a drug delivery tool
CPPs have been shown to be useful in transporting a variety of cargoes across the cell membrane and into the intracellular space.6,7,55,56 This ability means that CPPs hold great promise as efficient drug delivery tools. While the efficacy of transportation varies by CPP and cell type, CPP5s are capable of delivering peptides, proteins and nucleotides into most cell types once calibrated correctly. 55 This includes Leishmania tarentolae, a member of the order of Kinetoplastida that includes many species capable of causing serious human and animal disease. 57 CPP5 complexes were able to deliver both β-galactosidase and fluorescent-labeled bovine serum albumin with high efficiency despite the substantial difference in molecular weights between these compounds. 57 These studies suggest that CPP5s can be used to regulate eukaryotic as well as protozoa cells. However, as we reported previously, the cargo delivery activity is much lower than that of cationic CPPs such as TAT, and more than 10 µM concentration of CPP5 is necessary to achieve significantly detectable cargo delivery activity. Although a high concentration is necessary, CPP5s have the added advantage of no toxicity. 7 CPP5s may be useful for cell types that are sensitive to TAT and R8 (i.e. cells can be killed by TAT and other cationic CPPs). In particular, BIP has been shown to effectively transport peptides and proteins into cells, while also exhibiting antiapoptotic effects.7,58 This cytoprotective effect distinguishes BIP from other CPPs studied, which have shown dose-dependent cytotoxic effects. 7
Of note, BIP can be used to induce targeted gene expression in mesenchymal stem cells (MSCs). 59 A solid gold nanoparticle was coupled to CPP5 and used to induce enhanced expression of brain-derived neurotrophic factor (BDNF) in rat MSCs. 59 Within four days of a single exposure to the CPP5-nanoparticle complex, greater than 80% transfection was achieved. This mechanism of transfection holds promise in the development of therapeutic cell lines as part of the drive to deliver personalized medicine. The anti-apoptotic effect of BIP results in no loss of viability in transfection of these MSCs. This method of transfection has the potential to be both less expensive and less risky than transfection with viral vectors.
Recently, Wolfe et al. examined various CPPs for their efficiencies of nucleotide delivery into HeLa cells. 60 While the cationic CPPs such as TAT and poly-arginine (R8-R10) showed very strong activities, CPP5s showed very weak activities in delivering nucleotide in this study. This result is consistent with our previous report that CPP5's cargo delivery activity is weaker than that of cationic peptides.7,58 Although the weak cargo delivery activity is not attractive, the very low cytotoxicity (or even cyto-protective activity) of CPP5s is an advantage in its use as a drug delivery tool as discussed earlier. For example, the use of a high concentration of CPP (more than 0.1 mM) may overcomes the weak cargo delivery activity per each CPP5 molecule.
Recent progress in the pharmacological inhibition of Bax-mediated cell death (Figure 3)
In addition to BIP, there are several studies reporting the development of small compounds and antibody-derived peptides that inhibit Bax. We will review the representative inhibitors and discuss their potential as future therapeutics. Of note, Bak is another pro-apoptotic member of the Bcl-2 family of proteins which has a Bax-like activity to induce mitochondria-dependent programed cell death (reviewed in Korsmeyer 1 and Kale et al. 2 ). Bax is more ubiquitously expressed than Bak, and therefore, Bax plays a main role as the mediator of cell death in general. This is supported by the evidence that the Bak knockout mouse does not express a significant phenotype, while the Bax knockout mouse has increased neuronal survival and resistance to stresses activating mitochondria-dependent apoptosis. 61 Since Bax and Bak induce cell death by a similar mechanism, Bax inhibitors have been also tested against Bak and some of them showed dual inhibition of Bax and Bak as explained below.
Bax/Bak oligomer inhibitors
Nie et al. reported new five chemicals (molecular weight (MW) is ranging from 206–689) inhibiting the pore-forming activity of Bax. 62 These chemicals were selected from a group of 87 compounds that have binding affinity to Mcl-1, an anti-apoptotic member of the Bcl-2 family of proteins that shares Bcl-2 homology (BH) domains with Bax. The names of these compounds are BJ-1, BJ-1-BP, MSN-50, MSN-125, and DAN004. Compounds were initially screened via liposome dye release assay to determine whether they could inhibit Bax-mediated membrane permeabilization. IC50 values from the liposome dye release assay were found to be 9, 6, 6, 4, and 0.7 µM respectively. MSN-50, MSN-125, and DAN004 were found to be successful in inhibiting MOMP (mitochondrial outer membrane permeabilization) induced by Bax using isolated mitochondria in vitro. Since these inhibitors inhibited MOMP of bax−/−bak+/+ cells in addition to Bax proficient cells, the authors proposed that these inhibitors are Bax/Bak dual inhibitors. MSN-50 (5 µM) and MSN-125 (10 µM) inhibited apoptosis induced by actinomycin D and staurosporin (STS) in BMK (baby mouse kidney) cells. However, these chemicals were cytotoxic at concentrations of 20 µM and higher in the culture medium. In cultured primary embryonic mouse brain cortical neurons, MSN-125 (at 5 µM) also successfully reduced cell death if added immediately after glutamate excitotoxicity was induced (25 or 100 mM glutamate for 30 min). The authors propose that these new small molecules may be useful in many applications, but protection from traumatic brain injury was emphasized since these chemicals protected primary cultured neurons. The authors also discussed the unexpected off-target effects and issues surrounding the unknown mechanisms of Bax and Bak, which remain problems.
Bax channel inhibitor
Hez et al. describe two Bax channel inhibitors Bci1 (MW 423.67) and Bci2 (MW 395.6). 63 These chemicals were developed through the screening of a small chemical library to identify chemicals blocking the channel-formation by recombinant Bax proteins in the liposome. Bci1 and Bci2 inhibited fluorescent dye release from the liposome incubated with recombinant Bax protein, and the IC50 was 0.81 ± 0.22 and 0.89 ± 0.29 µM respectively. Release of the liposome’s contents was found to be near zero at 2.4 µM of both chemicals indicating that Bax was completely inhibited by this concentration. These chemicals were able to suppress cytochrome c release from isolated mitochondria at concentrations ranging from 10–20 µM. Furthermore Bci1 and Bci2 were able to inhibit STS-induced apoptosis in HeLa cells at concentrations of 5–10 µM in the medium. Evaluation of the inhibitors in an in vivo global brain ischemia gerbil model was also conducted. After ischemia induction, 3 and 30 mg/kg of Bci1 and 30 mg/kg of Bci2 were administered at 15 min, 24 h, and 48 h after reperfusion. After seven days, the doses of 3 and 30 mg/kg Bci1 was shown to have a reduction in hippocampal damage by 17% and 45%. Bci2 reduced damage by 55%. Cytosolic cytochrome c levels were also greatly reduced in animals that underwent the Bci1 treatment, which again suggests that Bax-mediated apoptosis was reduced. Hez et al. suggest that further improvement of these compounds could result in drug development to treat tissue damaged by ischemia/reperfusion injury.
Mitochondrial apoptotic channel inhibitors
Peixoto et al. reported on mitochondrial apoptotic channel inhibitors (iMACs),64,65 which are derivatives of di-bromocarbazole that have been shown to have anti-apoptotic effects in mouse models of neurodegenerative diseases and brain traumatic injury.66–69 In 2009, Peixoto et al., demonstrated that iMac2 (2.5 µM and higher) inhibited apoptosis in FL5.12 cells (derived from a murine, pro-B cell line) that were treated with STS or interleukin (IL)-3 deprivation. 64 They also showed that iMACs suppress cytochrome c release from mitochondria in these cells. In 2017, this group delved further into the exact mechanism of inhibition. 65 When treated with iMAC1 or iMAC2, FL5.12 cells subject to IL-3 deprivation showed a significantly decreased ratio of oligomeric to monomeric Bax compared with control cells. The authors propose that this may be due to disassembly of pre-formed MACs caused by treatment with the inhibitors. This theory is supported by the stepwise manner of inhibition seen in patch-clamping of mitochondria apoptotic channel (MAC) in giant liposomes through the addition of 5 nM tBid and 20–50 nM of recombinant Bax or Bak protein. It appears from these experiments that iMACs work both by blocking the initial translocation of cytoplasmic Bax to the outer mitochondrial membrane and by disassembling Bax oligomers already present. These inhibitors offer interesting insights into the mechanisms of mitochondrial permeabilization and apoptosis, and have promising utility in protection from pro-apoptotic stressors.
Utilization of Bax binding antibodies to inhibit Bax
Uchime et al. (2016) identified 14 novel synthetic antibody fragments (Fabs) capable of binding and inhibiting Bax. 70 The Fabs varied significantly in terms of complementary-determining region (CDR) sequences, and bound Bax at nanomolar concentrations, inhibiting the channel formation activity of Bax in the liposomal membrane. The diversity of CDR sequences found in these Fabs suggests significant variation in the mechanisms of interaction with Bax. The 3G11, a representative Fab, showed the binding activity to monomeric and cytosolic Bax. The 3G11 inhibited the translocation of Bax into the mitochondrial outer membrane. NMR and hydrogen-deuterium exchange mass spectrometry demonstrated binding of 3G11 directly to the N-terminal activation site. The authors suggest that 3G11 likely inhibited Bax translocation either by competition of truncated Bcl-2 interacting protein (tBID, a protein that activates BAX) or by preventing the conformational change required by Bax to translocate.
Robin et al. analyzed the structure and effects of inhibition of various sites on Bax to better characterize the activation process by which it translocates to the mitochondrial membrane. 71 In this study, authors also used an antibody (3C10) binding to the N-terminus of Bax. This study found that the 3C10 antibody inhibited cytosolic Bax from translocation via decreased exposure of the membrane-associated helix α9. This suggests that Bax inhibition works allosterically, by prohibiting Bax from forming its usual ensemble.
These synthetic antibodies are only for research purpose at present, however, these antibodies will provide the necessary information to develop clinically effective Bax inhibitors in the future.
Compound 22: A new mitochondrial permeability transition pore inhibitor
Fancelli et al. developed substituted cinnamic anilides capable of inhibiting mitochondrial permeability transition pores (mPTP). 72 The mPTPs are still not fully understood, but open in response to calcium overload and reactive oxygen species and likely contribute to the cell death seen in a variety of pathological states related to these conditions. 73 These cinnamic anilides reduced apoptosis by decreasing mitochondrial membrane permeability. The initial compound in this series of potent inhibitors, the 3-chlorophenyl of caffeic acid was originally identified through high-throughput screening of compounds capable of inhibiting swelling in rat liver mitochondria in response to calcium overload. In particular, compound 22 (one of these cinnamic anilides) was shown to have a dose-dependent and potent effect on the ability of mouse liver mitochondria to withstand calcium overload. This compound was significantly more effective than the standard mPTP inhibitor, cyclosporine-A (CsA). The authors determined that CsA and compound 22 likely interact with mitochondria in different ways, as the calcium retention capacity assay results plateaued at far lower concentrations in CsA (around 0.5 μM) than in compound 22. There was also an additive effect when compound 22 and CsA were used in combination. While CsA targets the cyclophilin D in mitochondria, compound 22 is thought to have a different target. In this study, compound 22 was shown to be capable of reaching and permeating rabbit cardiac mitochondria in an in vivo model. Rabbits were exposed to 30 min of left anterior descending coronary artery ligation followed by 4 h of reperfusion. Rabbits that received compound 22 just prior to reperfusion had a reduction in acute myocardial infarction size of 50%. These results suggest that inhibition of mitochondrial permeability can have a dramatic, positive effect on ischemia and reperfusion injury. Although the direct interaction of compound 22 and Bax has not been confirmed, the inhibition of mPTP is implicated in the protection of cells from Bax-mediated cell death at the point of mitochondrial cytotoxic calcium release.
Conclusions
Because of the ubiquitous expression of Bax in the human (and animal) body, therapeutically effective Bax inhibitors are expected to have a wide range of applications both in vivo and ex vivo. As we commented earlier, BIP has limitations in its use as a clinically effective therapeutic agent. Two representative limitations are the necessities of (1) a very high dose (more than 100 µM) and (2) a long pre-incubation time (2–16 h). Therefore, BIPs themselves may not be used as therapeutics at present forms. However, BIPs may be utilized as a drug delivery tool since BIPs are cytoprotective and much less toxic than other CPPs. Therefore, BIPs may have advantages over other mechanisms as a drug delivery tool. Several research groups have published the development of Bax-inhibiting small molecules that will become the basis of future therapeutics. Although these inhibitors are still at research use level, new clinically effective drugs protecting cells from Bax-induced cell death will be developed in the near future.
Footnotes
Authors’ contributions
All authors contributed the writing the manuscript.
DECLARATION OF CONFLICTING INTERESTS
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
FUNDING
This study was supported by research grants from NIH (RO1AG031903, Department of Defense (W81XWH-12–1-0331) and Foundation Fighting Blindness (FFB) (Gund-Harrignton Scholar Award).