
Abstract
Pharmaceutical drug development and clinical testing is associated with billions of dollars, and often the time and money spent does not result in a viable drug formulation. The pharmaceutical industry has long relied on animal models for testing efficacy, toxicity and specificity of novel drugs. However, the studies cannot be fully relied upon, as animal models are not reflective of human pathophysiology and drug response, which results in drugs being pulled from development as late as at stage IV, after billions of dollars have already been invested in such an effort. With the advent of adult-induced pluripotent stem cell technology, came an era which offered the potential of pursing human relevant developmental and pathogenesis research and drug testing on patient-induced pluripotent stem cell-derived differentiated cells, consciously reflecting human responses with regard to drug safety, toxicity, efficacy, and side effects. Specifically, human-induced pluripotent stem cell-derived hepatobiliary cells and tissues may be a more human-relevant model system to address the biggest barrier to drug safety and approval: hepatotoxicity. In this review, we address the potential of human-induced pluripotent stem cell-based hepatobiliary differentiation technology as a means to study human liver development and hepatic cell fate determination, and to model liver diseases in an effort to develop a new human-relevant preclinical platform for drug development.
Impact statement
In this review, we address the potential of human-induced pluripotent stem cell-based hepatobiliary differentiation technology as a means to study human liver development and cell fate determination, and to model liver diseases in an effort to develop a new human-relevant preclinical platform for drug development.
Introduction
Liver disease affects millions of patients worldwide. Many patients suffering from refractory liver diseases such as inherited metabolic liver diseases and end-stage liver failure may benefit from biologically active cellular therapy by either disease prevention or by treatment of the liver disease. Currently, the only treatment route available to patients with liver failure is an allogeneic liver transplant. 1 However, there is a shortage in availability of usable transplantable livers. To mitigate this, multiple avenues have been employed to expand the availability of donor organs including the use of suboptimal liver, split liver transplantation, liver transplantation from a living donor and opt-out organ donation programs.1–3 Despite these attempts, however, the demand for liver transplantation still greatly outstrips the availability of the organs. This prevents over 40% of patients on the transplant list from being able to be matched with a donor liver. 1 During the wait, these patients die before being matched or become too sick to be eligible for a transplant. Hence, there is an immense need for the development of new cellular therapies to reduce mortality and augment liver regeneration.
Amongst the alternative cellular therapies being utilized in lieu of whole liver transplantation, are focused on expansion of the available substitute liver tissues namely hepatocyte transplantation, engineered hepatic constructs, and the bioartifical liver system.1,4 Hepatocyte transplantation in particular has been suggested for treating acute liver failure and inherited metabolic liver disorder scenarios. 1 However, hepatocyte transplantation comes with its own set of disadvantages such as the limited engraftment potential, graft rejection. Furthermore, the challenge of sourcing appropriate donor cells is again in limited supply. 5 Despite, the innate potential of hepatocytes to regenerate and proliferate in vivo, in in vitro the efforts to recapitulate the same potential and induce proliferation of isolate human hepatocytes have proven unsuccessful. 5 Consequently, the search for alternative approaches to cell therapy brought about the use of hepatocellular carcinoma (HCC)-derived cell lines and SV40-transformed cell lines, which afforded the advantages of not only cell expansion but also the creation of in vitro model systems, but was also associated with disadvantages such as loss of hepatocyte function and acquisition of genetic abnormalities. 6 Alternative cell sources such as the use of fetal hepatocytes or xenogeneic materials have been suspended due to various sourcing, safety, and ethical considerations associated with them.1,7
In the case of disease treatment, the pharmaceutical industry has long relied upon nonhuman or animal models for testing the efficacy, toxicity, and specificity of novel drugs. However, not only are a majority of these animal-based studies costly, but furthermore they cannot be fully relied upon as, animal models cannot fully mimic human-specific biology/physiology, recapitulate disease development and phenotype, and further are not reflective of species-specific drug metabolism and response in humans. The pharmaceutical industry spends years and billions of dollars for drug development and testing. However, a report by the FDA showed that from every 100 drugs that passed animal testing successfully, 92 failed in human trials subsequently.8,9 For an investment of 10–15 years and a billion dollars, out of every 10,000 drugs that undergo research and further development, only 5–10 drugs progress to clinical trials, and out of these merely 1 will receive FDA approval. 10 Liver as the major organ of drug metabolism has a profound impact of drug effects, and hepatotoxicity is one of the main underlying causes for preclinical and clinical attrition of drugs. Primary human hepatocytes have been utilized to mitigate the disadvantages associated with animal models, but even these cells have disadvantages such as sourcing and aforementioned ethical considerations.
With the introduction of pluripotent stem cell (PSC) technology, came an era which offered the potential of not only developing an essentially replenishing source of hepatocytes for cellular therapies but also pursing disease modeling drug testing on human-induced pluripotent stem cell (iPSC)-derived differentiated functional cells, that consciously reflecting human in vivo responses with regard to drug safety, toxicity, efficacy, and side effects. Here, we address the potential of human iPSC-derived hepatobiliary cells/tissues as a means to study and model human liver diseases, capable of in vitro recapitulation of disease-specific phenotypes; and the utilization of these human disease models as a platform for screening and testing drugs, and for toxicity studies. Moreover, we discuss the potential of studying the cell fate determination process during hepatobiliary development using human iPSCs.
Liver disease modeling using human iPSCs
The differentiation of PSCs into varied cell types has been attempted both as a monolayer or as aggregates of cells. Embryoid bodies (EB) are formed from aggregates of PSCs, which can then be treated with precisely formulated combinations of cytokines and growth factors to induce specific lineage differentiation.11,12 Nonetheless, EB-based differentiation protocols have low efficiency in directed differentiation and are further complicated by spontaneous differentiation that results in development of unwanted cellular lineages. Differentiation of iPSCs in a monolayer has been utilized more widely, as it results in more homogenous cell populations than those obtained from the EB methods. In the monolayer-based approach for generating hepatocyte-like cells from iPSCs, protocols that mimic the embryonic liver development stages appear to be more promising.12–14 However, the efficiencies of these differentiation protocols are not constant across various research groups mainly due to cell line variation and the differences in culture conditions utilized. Much efforts have been made to optimize the process by excluding the utilization of less-defined cell culture elements such as animal serum, culture medium, animal feeder cells, and undefined extracellular matrix from animal tissues to promote consistent results with a higher efficiency.12–14
Highly efficient and functional protocols involving multiple differentiation stages and stage-specific differentiation have improved directed differentiation of human iPSC lines into hepatic lineage cells.12–14 The human iPSC differentiation technology has offered a highly attractive prospect and opens up a new avenue of toxicity screening using iPSC-derived healthy or diseased cells; which can be utilized before clinical trials to research drug safety, dosage, efficacy, response, and the effect of individual and population genetics in human cells.1,5 The application of human iPSC-derived liver cells for high-throughput compound screens could allow further offer insight into the underlying drug interactions and disease mechanism as successfully demonstrated in clinical drug screening using patient iPSC model of a liver disease. 15 This possibility of utilizing patient iPSC-based disease models has the potential to not only decrease the risk of failure linked to Phase I and II clinical trials but can also potentially help foster the development of precision medicine in terms of drug administration. 2
In addition, this human iPSC technology also allows us studying of the pathobiology of liver diseases under human-relevant biological conditions at the cellular and molecular level. Patient- and disease-specific iPSC-derived liver cells/tissue can now be leveraged to research and evaluate the molecular mechanisms underlying liver disease, progression, and treatment.2,4 Lately, many research papers have described the generation of patient- and disease-specific human iPSC lines and have further modeled these liver diseases.15–24 Disease- and patient-specific human iPSCs of glycogen-storage-disease, familial-hypercholesterolemia, and α-1-antitrypsin (AAT) deficiency liver disease have recapitulated important disease-specific pathological features and key disease phenotypes in vitro.2,15–17 Likewise, the restored function in the patient iPSC-derived hepatocytes in genetically corrected AAT-deficiency patient iPSC lines provides hope for future gene therapy.15,25 The successful derivation of disease- and patient-specific iPSC lines for other inherited metabolic liver diseases such as Crigler-Najjar syndrome, tyrosinemia, familial hereditary cholestasis, and glycogen storage disease has also been reported. 18 Wilson’s disease-specific human iPSC lines with the ATP7B gene carrying the R778L mutation have been reported to give rise to hepatocytes with flawed copper transport functionality in vitro. 24 The use of human iPSC-derived hepatocytes to model and evaluate alcohol-induced liver injury in vitro have reported that alcohol negatively influence liver progenitor formation and proliferation of hepatocytes.21,22 Interestingly, alcohol exposure appears to increase alcoholic liver disease associated phenotypes such as steatosis and HCC markers in the mature stage hepatocytes derived from human iPSCs. 21
More recently, human iPSC-derived hepatobiliary cells have been utilized to model biliary atresia and biliary fibrosis.26–28 These biliary disease-specific human iPSCs demonstrated two main biliary atresia-specific disease phenotypes, namely low efficacy in biliary differentiation and increased fibrosis. 28 When endoderm formation is transiently altered during normal liver development of human iPSCs, it gives rise to the development of abnormal profibrogenic proinflmmatory cholangiocytes, simulating those observed in biliary fibrosis patient livers such as biliary atresia and primary sclerosing cholangitis. 26
Human iPSC-derived hepatocytes have also been utilized to study hepatitis virus pathology 23 as well as malaria. 20 Patient-specific iPSCs from patients with HCC have been differentiated into hepatocytes instead yielding human hepatoma-like cells by knocking down cyclin-dependent kinase inhibitor 1 (p21). 19 Further, results showed a cyclic retinoid (10 µM) plus tolestat (10 µM) as an AKR1B10 inhibitor treatment would be an appropriate regimen for human hepatoma-like cells as a combination therapy. 19
Recently, research papers have reported the generation of human iPSCs from patients suffering from biliary diseases such as biliary atresia and cholangiopathies associated with other diseases27,29,30 and have started to be utilized in modeling these biliary diseases and drug discovery.29,30 Human iPSC-derived cholangiocytes from cystic fibrosis patients mimicked disease phenotype in vitro showing delayed cyst formation 29 or minimal CFTR protein expression. 30 Furthermore, polycystic liver disease patient iPSC-derived cholangiocytes have been generated and utilized to demonstrate the effectiveness of compounds that could help reduce cyst size, with octreotide (a synthetic somatostatin analog) reducing the organoid size. 30
Thus, human iPSC-derived liver disease models have shown that they hold great potential in studying pathophysiological development and pharmacological screening.
Modeling drug-induced liver toxicity using human iPSCs
Precise prediction of drug toxicity in humans is a crucial aspect of drug discovery and development process. Hepatotoxicity, especially, is one of the leading cause of drug failure during preclinical testing since a vast majority of drugs metabolize through the liver.10,31,32 Furthermore, the inconsistency observed in individual response to prospective therapeutic drugs, conferred by the inherent genetic differences is another major obstacle to overcome to improve the current drug development process. 33 And the inevitability of individual-specific adverse reactions to drugs has been becoming more and more apparent as new drugs of new classes are introduced. 34 The process of drug safety evaluation is further hampered by the quality, variability, and availability of primary human liver model systems available to better evaluate/predict drug toxicity.4,5
Human iPSC-derived hepatocytes have been employed to investigate toxicity of two compounds metabolized by either CYP2C9 or CYP2D6, where iPSC-derived hepatocytes were dosed with these compounds; followed by reduction in their viability, demonstrating the activation of these compounds into their reactive forms after CYP metabolism. 35 The CYP2D6 metabolized compound (BMS2) resulted in a near complete loss of iPSC-derived hepatocytes, while the CYP2C9 metabolized one (BMS1) resulted in only a nominal viability decrease. 35 This study demonstrated the feasibility of modeling toxicity in iPSC-derived hepatocytes that was comparable to the current gold standard used in industry, primary hepatocytes, and further showed the viability of modeling metabolic differences observed in a population by employing human iPSC-derived hepatocyte- like cells. 35
Valproic acid (VPA) toxicity has been modeled in Alpers-Huttenlocher syndrome (AHS) patient iPSC-derived hepatocytes, where the patients present with mutated POLG gene and with increased susceptibility to VPA-induced mitochondrial-dependent apoptosis and showed increased levels of activated Caspase-9 and cytochrome C release. 36 This study showed that in addition to the use of carnitine, which is capable of rescuing VPA-induced apoptotic sensitivity in iPSC-derived hepatocytes from an AHS patient, and has also been used for the treatment of VPA-induced liver toxicity in AHS patients, targeting the mitochondrial permeability transition pore (mPTP) may be an effective approach in preventing VPA-induced liver toxicity in AHS patients, as cyclosporine A (an mPTP inhibitor) rescued VPA-induced sensitivity to apoptosis in the patient iPSC-derived hepatocytes. 36 A research study aimed to compare the hepatic toxicity responses to pazopanib (PZ), a clinically prescribed drug for renal cell carcinoma treatment, in patients suffering from PZ hepatotoxicity, as compared to patients who received PZ but saw no such hepatotoxic effects. 37 Transcriptome analysis revealed that the mechanism through which PZ induces damages is through oxidative stress. Further transcriptomic comparative analysis revealed proof of variable iron metabolism and protracted oxidative damage in susceptible iPSC-derived hepatocytes, which may account for the differences seen in individual hepatotoxic responses seen to PZ treatment. 37 Human iPSC-derived hepatocytes have also been utilized to screen a library of 240 drugs, and then subjected to multi-fluorescent imaging, which demonstrated Calcein AM and MitoTracker Orange as the most sensitive markers of viability. 38 This study provides the tools required for the characterization of multiple phenotypes of toxicity using live cells.
Hepatocytes derived from human iPSC lines, thus, present an innovative and a more human-relevant platform to mimic and study the inherent idiosyncratic hepatotoxicity and disease-specific features, and further allows for the simultaneous examination of the mechanisms underlying these unique toxicity responses with numerous intrinsic elements at play, in appropriate patient-specific landscapes.35–38 Furthermore, taking advantage of highly improved and accurate gene editing technologies,25,39,40 hepatocyte banks from a genetically diversified panel of human iPSCs may very well be extremely valuable in being able to represent genetic diversity of a population in the context of drug testing and toxicity screening.
Potential of human iPSCs in dissecting human liver development process and the related key signaling pathways
Liver development process has been largely studied using animal models.41,42 More efficient and functional differentiation protocols have been developed to improve the hepatobiliary differentiation of human iPSCs, that involve multiple stages, and a stage-specific step-wise differentiation schema mimicking the human hepatobiliary development.12–14,43 Thus, iPSC-based hepatobiliary differentiation will provide a critical human-relevant biological tool to study the key signaling pathways associated with liver development that have previously been discovered using animal models.
The Hippo signaling pathways is one of the important molecular pathways in liver development, originally described in the fruit fly, Drosophila melanogaster, where the loss of the Hippo pathway enlarged its organs due to excessive proliferation and decreased apoptosis. 44 In addition, in conditional knockout animal models of YAP, a vital protein in the Hippo signaling pathway in cholangiocytes and hepatocytes, it was shown that though the ductal plates formed normally, the process of bile duct tubulogenesis fails, leading to bile duct paucity two weeks post-natal. 45 In adult liver, this bile duct paucity in turn induces a ductular reaction joined by steatosis and fibrosis. In contrast to its critical role in liver development, YAP1 activity does not seem essential in quiescent adult liver. 46 However, it plays a key role during liver regeneration, and its activity is required post liver injury to generate sufficient proliferation.46,47 Bile duct ligation (BDL) in mice induces a cholestatic injury which further triggers reparative responses such as proliferation of hepatocytes and cholangiocytes. 48
In adult mouse liver, bile ducts show high expression levels of YAP, with particularly robust expression in cholangiocytes. 45 In contrast, hepatocytes showed low levels of YAP expression. The loss of LATS1/2, the direct upstream regulators of YAP and TAZ, activated YAP/TAZ complex during and post liver development. YAP/TAZ activation repressed HNF4A expression, which in turn suppresses the differentiation of bi-potent hepatoblasts into hepatocytes, and instead promotes the commitment of hepatoblasts to the biliary epithelial cell type. 49 Furthermore, studies have reported ectopic YAP expression in mature hepatocytes, leads to de-differentiation of the mature hepatocytes into biliary ductal/progenitor cells, which can again differentiate back into hepatocytes after YAP activity levels decrease.50,51
Another morphogenetic pathway critical for liver development and in oncogenesis is the Hedgehog (Hh) signaling pathway.52,53 Hh and its downstream regulator Gli1 are expressed during fetal liver development, in the ventral foregut endoderm, from which the liver bud develops, and their expression is sustained even in the advanced liver developmental stages. 52 Regeneration or the reparative responses in the liver are accompanied by a reactivation of this signaling cascade, and alterations of Hh signaling activity have been observed in cholangiopathies like biliary atresia, 54 primary biliary cholangitis and Meckel-Gruber syndrome.55–57 The Hh pathway plays a vital role in cholangiocyte pathogenesis, as shown in a cholestatic injury model.56,58 In rodents with BDL, the expression of Hh ligands in the liver and the upregulation of Hh pathway has been observed to dramatically increase. Furthermore, in mice with genetically ablated patched, the transmembrane Hh receptor showed aggravated ductular and fibrogenic responses.
All these studies were largely based on animal models and/or limited human tissue sections which do not allow biological or functional assays. Therefore, human iPSC-based multistage hepatobiliary differentiation technology will be greatly useful to dissect the biological roles of YAP, Hh, or other important signaling molecules in each stage of human liver development. Based on the hepatobiliary differentiation cultures of human iPSCs, the expression of YAP1 increases along with both hepatocytic and biliary differentiation processes of human iPSCs, and the levels were significantly increased in the iPSC-derived biliary cells compared to the hepatocytes derived from iPSCs (Figure 1). This suggests the potentially important roles of YAP in human liver development, especially in biliary commitment and maturation. Thus, iPSCs may potentially provide a new human relevant biological tool to study the important signaling and cell fate changes in normal or abnormal liver development.
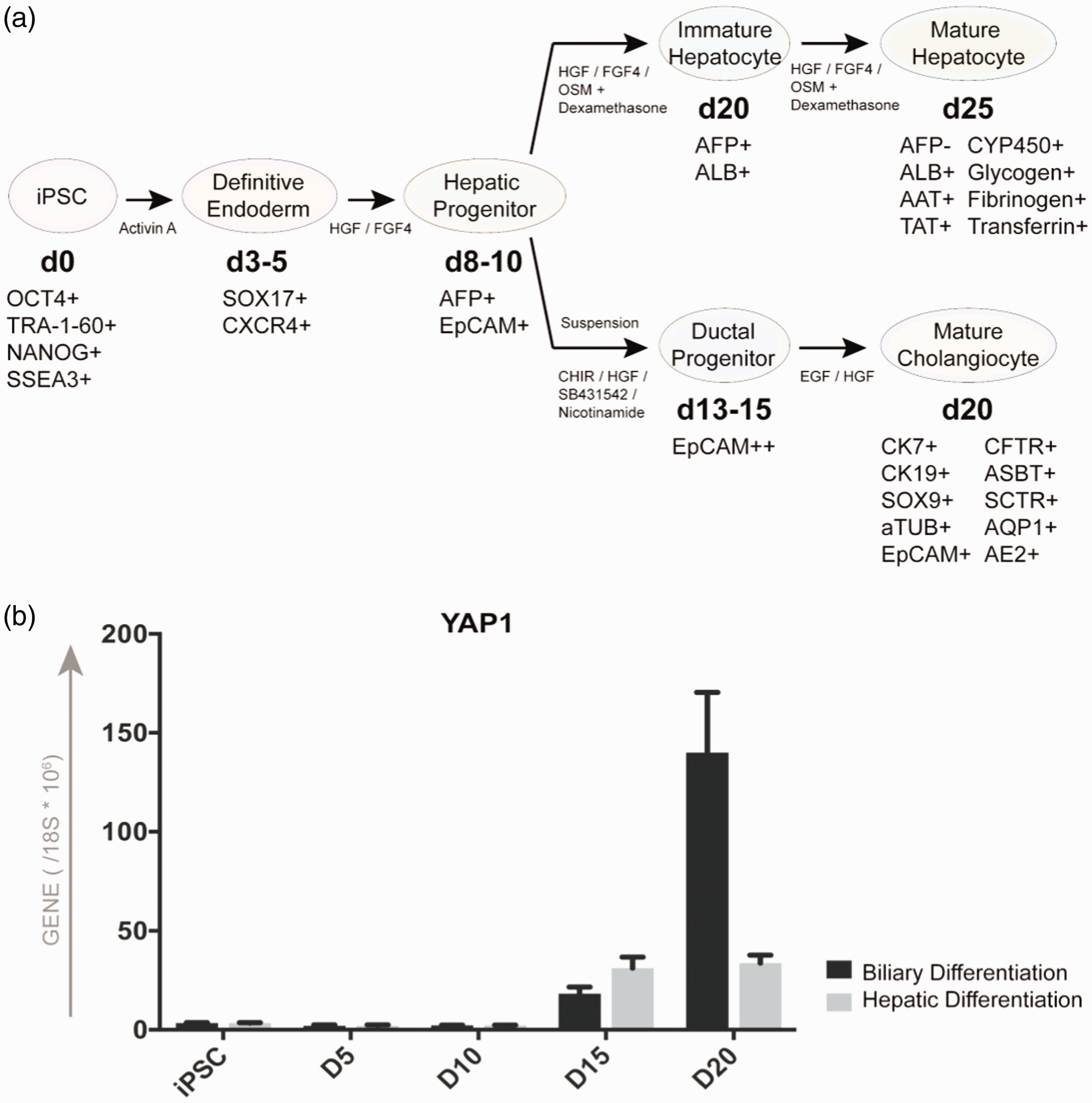
YAP expression changes along with multistage differentiation of human iPSCs into hepatobiliary epithelial cells. (a) Human iPSCs can differentiate into definitive endoderm by the application of activin A, and further into the hepatic progenitor cells under the influence of HGF and FGF4. Hepatic progenitor cells differentiate into hepatocyte-like cells when cultured in hepatocyte culture medium when supplemented with HGF, FGF4, OSM, and dexamethasone. Hepatic progenitor cells can also be induced to form ductal progenitor cells in medium supplemented with CHIR, HGF, SB431542, and nicotinamide. These ductal progenitors differentiate into cholangiocyte-like cells in biliary culture medium supplemented with EGF and HGF. (b) Kinetics of quantitative expression of YAP1 in directed hepatocytic and cholangiocytic differentiation of human iPSCs which are shown in (a).
Conclusion
In conclusion, human iPSC-derived hepatobiliary tissues show an immense potential not only as a seemingly unlimited source of hepatic tissues for therapies involving cell replacement but also provide an essential avenue to study normal and abnormal human liver development on an unprecedented level in a human-relevant biological setting, and for dissecting the molecular mediators and signaling pathways governing it. Furthermore, the technology offers up the opportunity to investigate the evolution and pathogenesis of acute and chronic human liver diseases, and to tease out the critical molecular mediators driving disease inception and progression. Human iPSC-derived hepatobiliary tissues further provide a resource to model liver diseases that recapitulate disease phenotypes in vitro which can be utilized for drug screening, testing, and toxicity studies to provide a more human-relevant and accurate ADME, drug toxicity, and safety information.
Footnotes
Authors’ contributions
Y-YJ: study conception and design, PC, LT: performed experiments, ZY: provision of study material, PC, ZY, LT, Y-YJ: data analysis and interpretation, PC, Y-YJ: wrote manuscript, Y-YJ: obtained funding.
DECLARATION OF CONFLICTING INTERESTS
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
FUNDING
This work was supported by NIH grants R01EB023812 and R03HD091264, and Johns Hopkins Pediatric Liver Center, Johns Hopkins Children’s Center. Funders had no involvement in study design, interpretation or decision to publish.