
Abstract
The process of cancer development and progression is driven by distinct subsets of cancer stem cells (CSCs) that contribute the self-renewal capacity as the major impetus to the metastatic dissemination and main impediments in cancer treatment. Given that CSCs are so scarce in the tumor mass, there are debatable points on the metabolic signatures of CSCs. As opposed to differentiated tumor progenies, CSCs display exquisite patterns of metabolism that, depending on the type of cancer, predominately rely on glycolysis, oxidative metabolism of glutamine, fatty acids, or amino acids for ATP production. Metabolic heterogeneity of CSCs, which attributes to differences in type and microenvironment of tumors, confers CSCs to have the plasticity to cope with the endogenous mitochondrial stress and exogenous microenvironment. In essence, CSCs and normal stem cells are like mirror images of each other in terms of metabolism. To achieve reprogramming, CSCs not only need to upregulate their metabolic engine for self-renewal and defense mechanism, but also expedite the antioxidant defense to sustain the redox homeostasis. In the context of these pathways, this review portrays the connection between the metabolic features of CSCs and cancer stemness. Identification of the metabolic features in conferring resistance to anticancer treatment dictated by CSCs can enhance the opportunity to open up a new therapeutic dimension, which might not only improve the effectiveness of cancer therapies but also annihilate the whole tumor without recurrence. Henceforth, we highlight current findings of potential therapeutic targets for the design of alternative strategies to compromise the growth, drug resistance, and metastasis of CSCs by altering their metabolic phenotypes. Perturbing the versatile skills of CSCs by barricading metabolic signaling might bring about plentiful approaches to discover novel therapeutic targets for clinical application in cancer treatments.
Impact statement
This minireview highlights the current evidence on the mechanisms of pivotal metabolic pathways that attribute to cancer stem cells (CSCs) with a special focus on developing metabolic strategies of anticancer treatment that can be exploited in preclinical and clinical settings. Specific metabolic inhibitors that can overwhelm the properties of CSCs may impede tumor recurrence and metastasis, and potentially achieve a permanent cure of cancer patients.
Introduction
To date, many lines of research suggest that cancer can be considered as a metabolic disease characterized by defective respiration or enhanced glycolysis. Genomic alterations and other hallmarks of cancers are nowadays conceived as downstream epiphenomena of underlying causes of metabolic disorders in cancer cells. 1 Dysregulation of glucose metabolism, fatty acid synthesis, and glutaminolysis that satisfy the bioenergetic and biosynthetic needs of cancer cells can cause their resistance to therapies. 2 As such, it is evident that cancer cells can modulate their metabolism to optimize energy demands 3 and induce immune escape, 4 which could be exploited as therapeutic targets for cancer treatment in light of the evolving metabolic landscape. Given that cancer stem cells (CSCs) embrace many malignant traits conferring therapeutic resistance, recurrence, and metastasis, eradication of CSCs can hold a promise of permanent cure for cancer patients.
CSCs manage to retain stem cell properties embracing the capacity of self-renewal and differentiation into progenies to form the epigenetically defined intra-clonal bulk tumors. It is reminiscent of normal stem cells and transit-amplification of cell populations with differentiation capability,5,6 which is called “stemness” of cancer cells. Metaphorically, the majority of heterogeneous tumor cells resemble various kinds of worker bees and CSCs are like the queen bees that establish the bee colony and heterogeneity of the hierarchy. Without the queen, the whole colony will collapse. Likewise, CSCs sustain the entire cancer population in a similar fashion. Unfortunately, conventional cancer therapies kill only differentiated tumor cells but spare CSCs, an extremely rare subset of resistant cells within the tumor mass, which is the main cause of recurrence and metastasis. 7 This is due to the fact that CSCs are able to expel anticancer drugs via ABC-transporters. 8 Peculiar properties of CSCs, such as manifesting quiescent phenotype, maintaining reactive oxygen species (ROS) at low levels, and residing in hypoxic region within tumors may also compromise the effectiveness of anticancer therapies. 9 These underlie the observation that CSCs are refractory to treatment and have a high incidence of relapse even after therapy has almost eradicated the tumors. Finding CSCs may provide an effective direction and a major leap toward a cure of cancers. Spherogenesis, side population, cellular markers, and aldehyde dehydrogenase (ALDH) activity of CSCs are currently major avenues to characterize and isolate CSCs. 10 However, there are a lot of challenges of isolating and enriching such rare CSCs from heterogeneous tumor bulk in the absence of knowing their specific biomarkers. An alternative characterization of CSCs from a metabolic perspective provides a new direction to identify and target this rare subpopulation. Therefore, the metabolism-based therapy has become a dawning strategy to halt cancer progression, and substantial efforts have been made to discover novel targets for anticancer treatment. In this review, we highlight the key players of metabolic pathways that characterize CSCs with insights in metabolic therapeutic strategies that can be exploited in preclinical and clinical settings. Specific metabolic inhibitors that can overwhelm stem cell properties may halt disease recurrence and attenuate the dissemination of CSCs and the capacity of spawning metastases of cancer cells.
Metabolic reprogramming in the CSCs
The metabolic hallmarks of CSCs in different types or status of tumors like proliferative vs. quiescent or normoxic vs. hypoxic have been mired in controversy, and the molecular mechanism governing the stemness properties of CSCs remains unclear. 11 Differences in tissue oxygenation and genetic background may be one of the causative factors in the development of metabolic complexity, plasticity and heterogeneity in different types of CSCs. 12 Remaining controversies may be related to diverse surface markers and isolation techniques of CSCs in different tumor types. 13 Notwithstanding the divergent findings in the metabolism of CSCs, a corpus of evidence is emerging to show that these unique metabolic signatures are pivotal to the function of CSCs. The discoveries of stem cells spark heated debate over whether CSCs employ central metabolic pathways as seen in normal stem cells, which are quite dissimilar to the differentiated bulk of tumor cells. Even though bioenergetic signature of CSCs has remained elusive, many lines of evidence have accumulated to demonstrate that metabolic pathways in CSCs are like those in tissue stem cells. The metabolic signature of stem cell-like osteosarcoma resembles that of somatic stem cells characterized by an upregulation of glycolytic flux. 14 This metabolic shift of CSCs encompasses a decrease in oxidative phosphorylation (OXPHOS) but a pronounced increase in lactate production, expression of glycolytic enzymes and glucose consumption compared with their differentiated counterparts. Although glycolysis is enhanced in the CSCs of osteosarcoma, 14 breast cancer, 15 nasopharyngeal, 16 colorectal, 17 and hepatocellular carcinoma, 18 many studies have shown that CSCs of glioma, 19 pancreatic ductal adenocarcinoma, 20 leukemia 21 rely mainly on mitochondrial respiration for the major supply of energy (Table 1). These oxidative CSCs are less glycolytic and have increased levels of mitochondrial mass, membrane potential, ATP, and ROS concentrations as compared with their differentiated counterparts. Pancreatic cancer cells with oncogene ablation acquired stemness traits to display active mitochondrial function and rely less on glucose and glutamine metabolism, but more on pyruvate and palmitate to fuel the tricarboxylic acid cycle. 22 However, the reliance on glycolysis or OXPHOS is still ambiguous in glioblastoma, lung, breast, and ovarian CSCs. These diverse observations may stem from the fact that the definition and isolation methods of CSCs were not identical among these studies, and hence different CSCs came from a variety of sources with divergent characteristics. In any case, metabolic heterogeneity of different subclones of CSCs can bear different metabolic phenotypes because of differences in genetic or microenvironmental factors of cancers (Figure 1).13,23
Cancer stem cells with different metabolic characteristics in various cancer types.
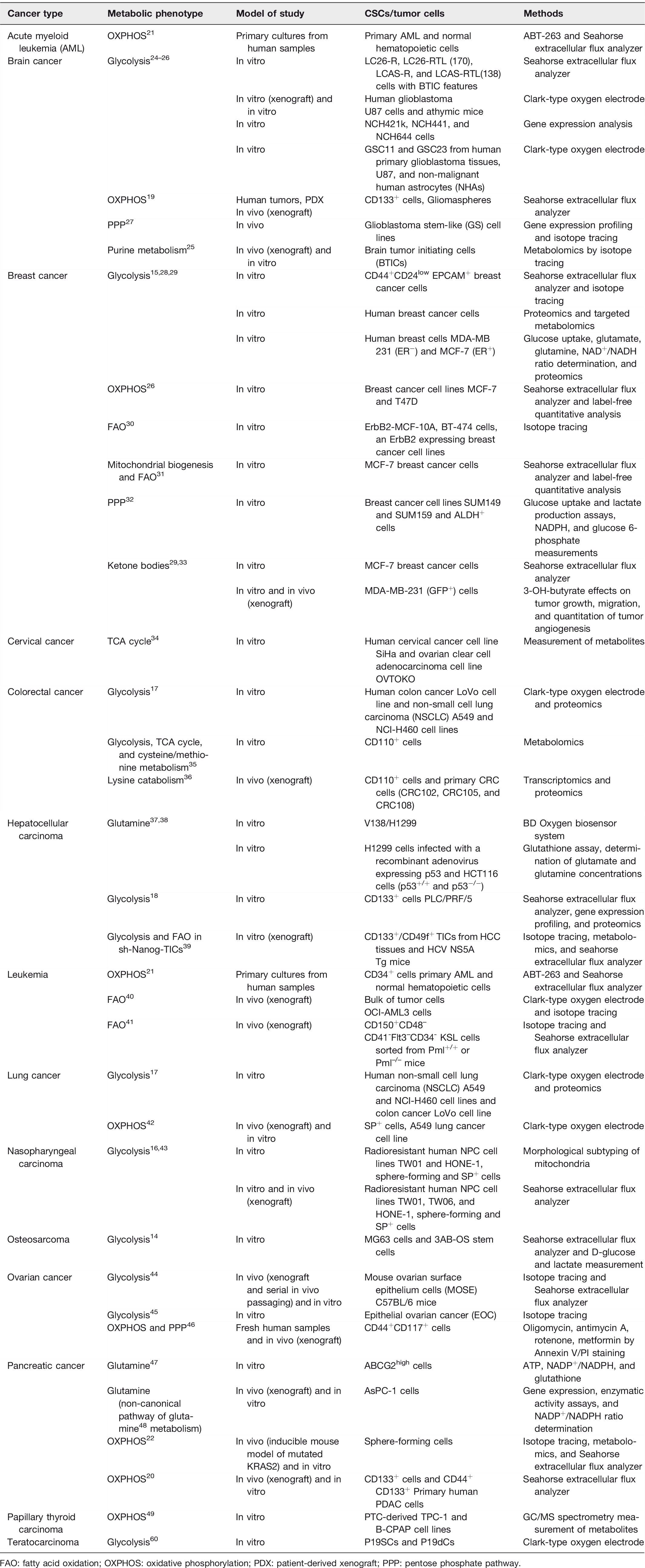
FAO: fatty acid oxidation; OXPHOS: oxidative phosphorylation; PDX: patient-derived xenograft; PPP: pentose phosphate pathway.
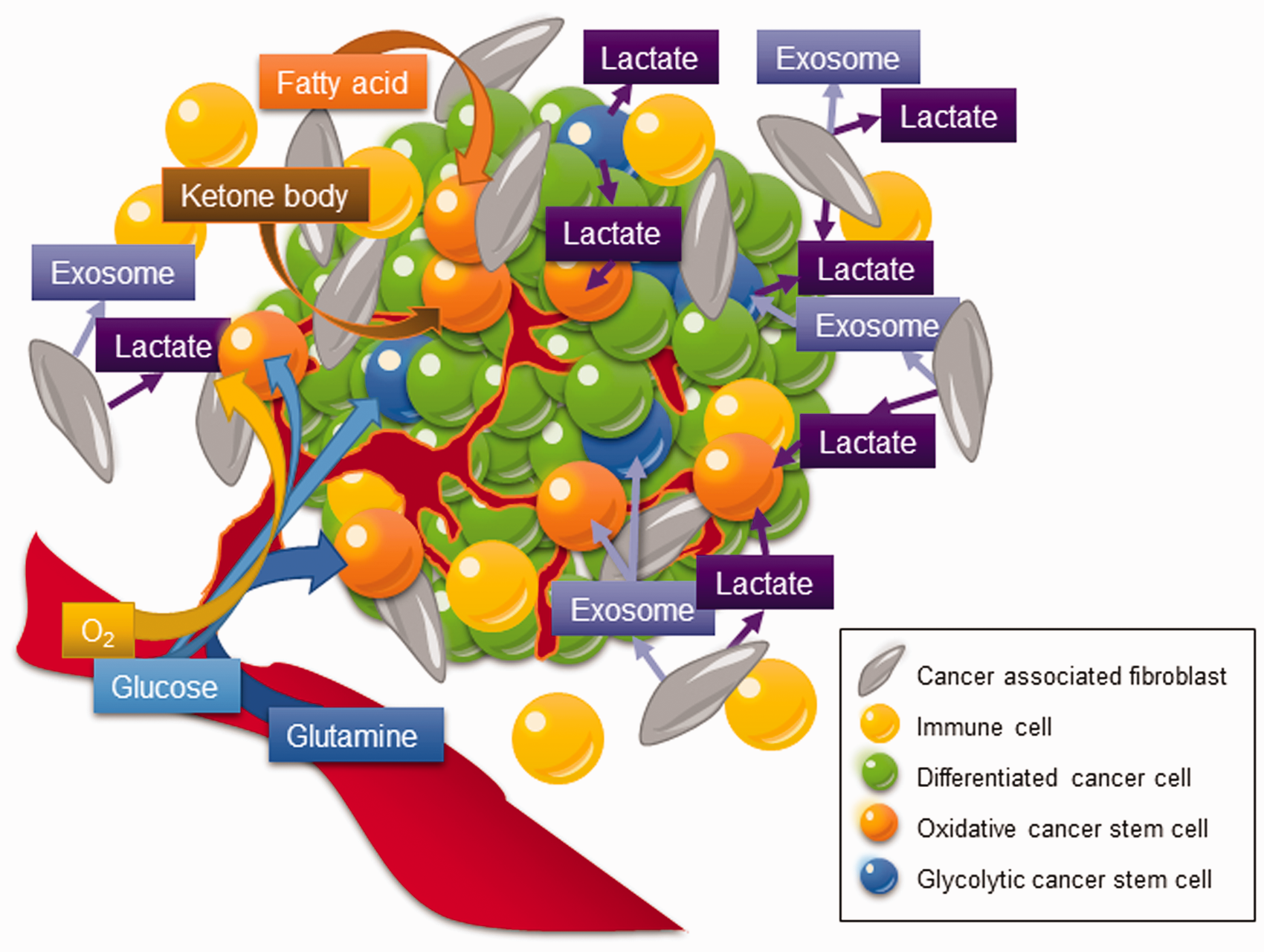
Metabolic heterogeneity of CSCs. Instead of dwelling in a solitary place, CSCs reside in a diversified ecosystem encircled by extracellular matrix, endothelial cells, immune cells, and cancer-associated fibroblasts (CAFs). In contrast to their differentiated counterparts, CSCs display an exquisite metabolic characteristics according to the type of cancer, which may predominately rely on glycolysis, catabolism of glutamine, fatty acids, and certain amino acids. Heterogeneity of CSC metabolism across different types and microenvironment of tumors confers CSCs to have the plasticity to cope with the endogenous mitochondrial stress and/or exogenous microenvironment. (A color version of this figure is available in the online journal.)
In addition to glucose metabolism, recent reports have disclosed the peculiarity of CSCs metabolism including lipid metabolism, redox state, and utilization of alternative fuels including amino acids and ketone bodies (Figure 1 and Table 1). Leukemia-initiating cells and hematopoietic stem cells may depend on mitochondrial fatty acid oxidation (FAO) for the production of ATP and NADPH.30,41 Glioblastoma CSCs can switch from higher pentose phosphate pathway (PPP) activity for elevated proliferation rate under acute oxygenation to a glucose-dependent status under hypoxia and a migrating process to protect cells against hypoxic cell damage. 27 Besides, oxidative stem-like cells may be fueled by more differentiated glycolytic somatic cells through a “reverse Warburg effect” under normoxic conditions in breast cancer. 51 Epithelial-mesenchymal transition (EMT) contributes stemness to CSCs with the capabilities to catabolize pyruvate, lactate, amino acids such as glutamine, glutamate, and alanine, as well as ketone bodies from the microenvironment to support mitochondrial respiration in CSCs. 52 Intriguingly, tumor cells with impaired respiration can regain mitochondrial function by receiving mitochondrial DNA (mtDNA) from the surrounding cells in tumor microenvironment, which renders tumor-initiating capacity and therapeutic resistance.53,54 Cancer-associated fibroblasts (CAFs)-derived exosomes are laden with mtDNA, which can be released into CSC niches and consumed by dormant CSCs. 55 This horizontal transfer of mtDNA promotes respiration-proficient CSCs with higher OXPHOS potential and the development of hormonal therapy resistance. 55 The horizontal transfer of mtDNA recapitulates the pivotal metabolic interaction between CSCs and the tumor microenvironment (Figure 1).
As for the mitochondrial distribution, the mitochondria are widely scattered over the cell periphery in well-differentiated cells, whereas these organelles are clustered in a peri-nuclear fashion in CSCs. 16 Similarly, the mitochondria are peri-nuclear clustered in embryonic stem cells (ESCs) and are rearranged to disperse throughout the cytoplasm upon differentiation. 56 The peri-nuclear accumulation of mitochondria supports the transportation of mitochondrial transit peptide to strengthen mitochondrial function. 57 It may also indicate that ATP would be efficiently transported from mitochondria to the nucleus, which is refrained from fluctuations in Ca2+ levels that often occur in the cytoplasm. 57
Likewise, reduction-oxidation (redox) homeostasis and mitochondrial membrane potential (Δψm) are imperative hallmarks for a balance between differentiation and sustaining stemness status of stem cells and CSCs.58–60 In contrast to differentiated tissue cells, normal hemopoietic and mammary epithelial stem/progenitor cells reside in a low oxygen niche, that is essential to self-renew and deter from differentiation.58,61 Stem cells launch the expression of antioxidant enzymes to maintain ROS at low levels for sheltering the cells from oxidative damage. 62 Oppositely, raising the ROS levels can compel stem cells to a lineage-specific differentiation. 63 In conformity with normal stem cells, CSCs with augmented antioxidant defense systems keep ROS levels at bay and may confer CSCs with survival advantage and therapeutic resistance.9,16,46 Since mitochondrial respiration is a notorious intracellular ROS sources, preferential dependence on glycolysis in certain types of CSCs is considered a way to shield themselves from accumulating too much ROS. Glycolytic pathway can consolidate the antioxidant defensive mechanism through generating NADPH by glucose 6-phosphate dehydrogenase (G6PD) for the recycling of the low molecular weight antioxidants including GSH (reduced glutathione), thioredoxin and peroxiredoxin through glutathione reductase. It was shown that ROS generation results in mitochondrial depolarization, 64 inferring that CSCs with low ROS levels may retain the mitochondrial membrane integrity and normal Δψm. Impairment or downregulation of mitochondrial uncoupling proteins may also result in low ROS but high Δψm levels, which sustain the malignancy of CSCs. 60 The Δψm is also regarded as a prerequisite factor for ESCs to differentiate or initiate teratoma formation. 65 ESCs with a high Δψm tend to develop teratoma, while a decrease of the Δψm in ESCs is able to stagnate tumor growth. 65
In essence, it seems that the CSCs are equipped with an extraordinary bioenergetic machinery to cope with oxidative stress, hypoxia, and challenge of the microenvironment. This dynamic and multidimensional Warburg effect of CSCs compared with differentiated tumor counterparts is specifically referred to as “the Warburg effect 2.0” to signify the upgraded and pellucid version of CSC pathophysiology regarding the CSC heterogeneity across tissue-specific variations, distinct histologic tumor subtypes, as well as the CSCs niche. 66
Metabolic regulators wheel CSC properties
Accumulating evidence reveals that pluripotency transcription factors such as NANOG, MEIS1, Wnt, or OCT4 are inextricably intertwined with metabolic regulators such as MYC, p53, K-Ras, and HIF1α, and their link with metabolic reprogramming dictates the stemness properties of CSCs.67,68 Certain metabolic signatures of CSCs act as a coordinator of stemness to perform several intricate functions in sustaining the stemness characteristics. Therefore, we highlight the therapeutic potential of these critical metabolic regulators that may govern the metabolic plasticity and redox homeostasis in CSCs (Figure 2). 68
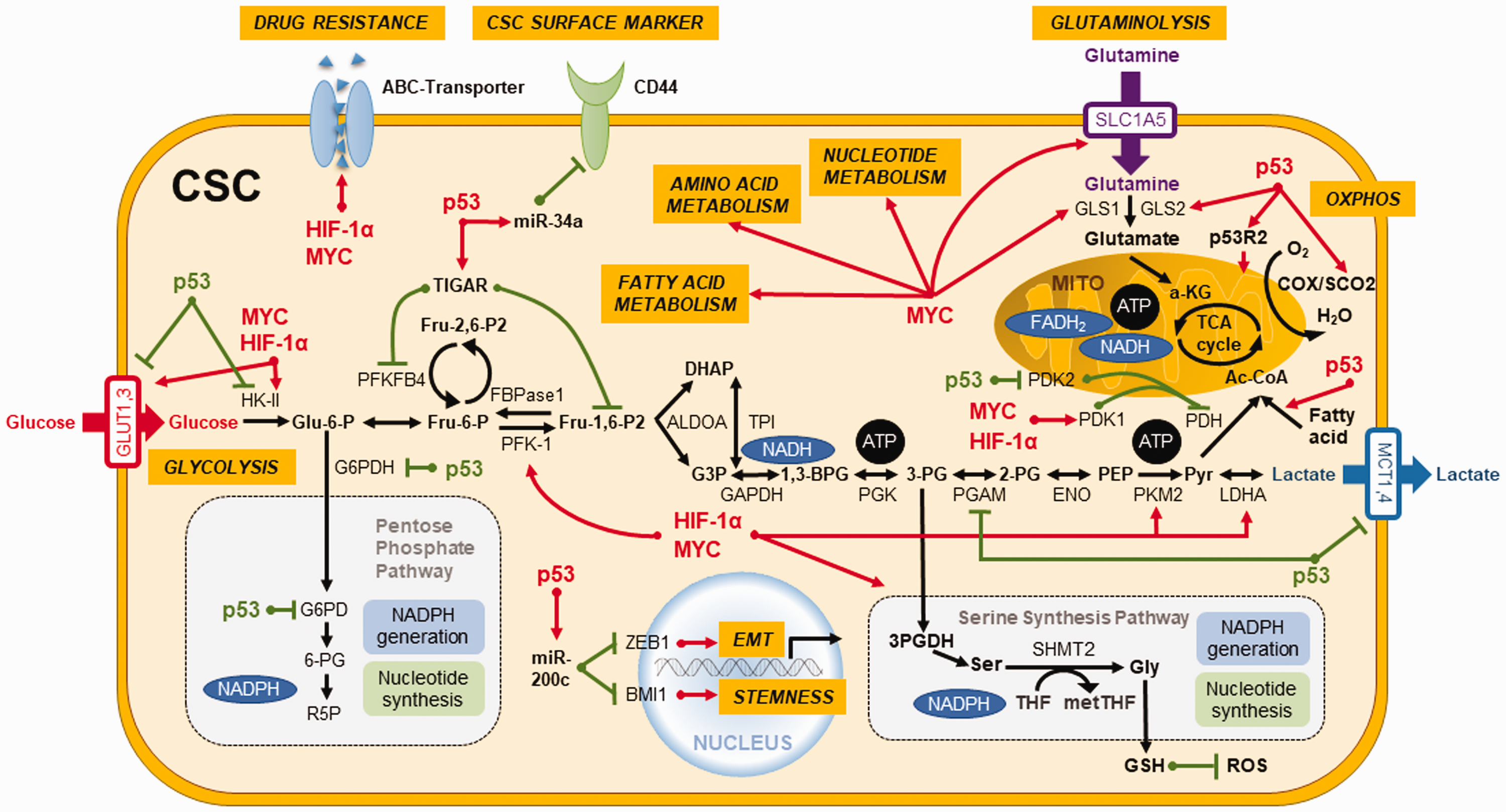
Key regulators orchestrate metabolic plasticity and stemness features in CSCs. Critical transcription factors such as MYC, HIF-1α, and p53 not only govern metabolic plasticity and redox homeostasis in CSCs but also serve as coordinators in regulating the stemness traits and differentiation capabilities of CSCs. Activation is represented by red lines and suppression is denoted by green lines. (A color version of this figure is available in the online journal.)
p53
TP53 is an extensively studied tumor suppressor gene in oncology research. 69 Occurrence of p53 mutations represents an early event in more than 50% of human malignancies. 70 p53 plays a crucial role in regulating glycolytic pathway through TIGAR (TP53-induced glycolysis and apoptosis regulator) and phosphoglycerate mutase (PGM) to suppress glycolytic enzymes and downregulate the transcriptional levels of glucose transporter 1 (GLUT1) and GLUT4 (Figure 2). 71 Additionally, p53 upregulates mitochondrial metabolism by enhancing the biosynthesis of synthesis of cytochrome c oxidase 2 (SCO2) and glutaminase 2 (GLS2) (Figure 2).37,72 Also, p53 abrogates the biosynthesis of nucleotides and lipids in tumor cells by attenuating the PPP (Figure 2). 73 Moreover, p53 reduces the biosynthesis of fatty acids but boosts FAO, thereby promoting OXPHOS by the increase of metabolic products of FAO in mitochondria of cancer cells (Figure 2). 74 In 2007, it was reported that adult human fibroblasts can be reprogrammed into ESC-like induced pluripotent stem cells (iPSCs) by overexpressing defined factors. 75 Upregulation of glycolytic genes was unveiled to precede the expression of stemness markers in the reprogramming process. 76 In addition, p53 pathway attenuates the reprogramming efficiency of somatic cells to iPSCs 77 and CSCs. 78 The absence of p53 imparts the resistance of colon CSCs to paclitaxel because of increased autophagy and decreased apoptosis. 79 Moreover, p53 was found to upregulate miR-34a to dampen the expression of CD44, which is a CSC surface marker that participates in the metastasis of CSCs. 80 In addition, p53 is able to upregulate miR-200c to reverse the stemness and EMT of CSCs (Figure 2). 81 We demonstrated that the reactivation of p53 by resveratrol could impede the stemness, EMT process, and metabolic remodeling of CSCs in nasopharyngeal carcinoma. 43 Taken together, p53 may be considered as an important target for CSCs-targeting therapy.
MYC
MYC regulates cancer metabolism and redox balance by boosting glycolysis 82 and glutaminolysis by upregulation of glutamine transporters and mitochondrial GLS through microRNAs (Figure 2).83,84 Glutamine fuels ATP production and intermediates in the biosynthesis of amino acids, nucleic acids, fatty acids, and glutathione, which bears powerful antioxidant and other biological activities. 85 In line with the findings in iPSCs, overexpression of MYC acts as a bridge between glycolysis and stemness in cancers. 67 MYC upregulates stemness and differentiation, as well as signaling pathways that result in the chemotherapy resistance of CSCs.67,86 Silencing of c-MYC was found to re-sensitize colon CSCs to chemotherapeutic agents through down-regulation of ABC transporters (Figure 2). 87 In CSCs, metabolic reprogramming driven by MYC is linked to CD44 variant isoforms (CD44v)-mediated redox homeostasis, tumor suppressor FBW7-driven c-MYC degradation, and ubiquitination, which are associated with chemoresistance. 88
HIF
HIF-1α and HIF-2α have a pivotal role in cancer cell progression for CSCs to trigger the metabolic reprogramming from an oxidative to a glycolytic phenotype to cope with low levels of oxygen, pH, and nutrients in the stringent tumor microenvironment (Figure 2). 89 HIF-1α regulates cancer migration, angiogenesis, and cell survival pathways, 90 and it also upregulates carbonic anhydrases for controlling pH gradient between intracellular and extracellular environments.91,92 This pH shift affects the drug absorption and metabolism and suppresses cytoplasmic retention of anticancer drugs. 93 Overexpression of HIF in CSCs enhances cancer progression through upregulation of PKM2 (pyruvate kinase muscle isozyme), ABC transporters (Figure 2), vascular endothelial growth factor, angiogenesis, recruitment of tumor-associated macrophages, and CD8+/− T cells, as well as attenuation of natural killer cells.94–96
Other metabolic regulators
Aside from a rapid ATP generation, glycolytic flux can supply metabolites to furnish PPP to produce NADPH and biosynthetic building blocks to support anabolic processes and protect CSCs from oxidative stress. 97 In breast CSCs, glucose transporters GLUT1 and GLUT4 and key glycolytic enzymes including HK (hexokinase), PKM2, and lactate dehydrogenase A (LDHA) control the central pathway of glucose catabolism and display increased activities. However, treatment with 2-DG (2-deoxyglucose), a glucose analogue that suppresses HK2, preferentially interferes with the viability of CSCs compared with differentiated tumor counterparts, demonstrating the crucial roles of glycolysis in the maintenance and growth of breast CSCs. 98 Additionally, PKM2 and PFKFB4 (6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4) were corroborated to be critical stemness regulators in glioma stem cells. 99 The interaction of nuclear PKM2 and OCT4 can upregulate stemness genes to enrich the subset of CSCs under metabolic stress and thereby promote cancer metastasis. 100 LDHA generates lactate to adjust pH and direct USP28-mediated de-ubiquitination and stabilization of MYC and activation of SLUG promoter, which endows breast cancer cells with stemness features. 101 Inhibition of G6PD, the gatekeeper of the PPP, decreased spherogenesis and the ALDH activity in breast CSCs. 32 Besides, phosphorylation of Bcl-2 associated death promotor (BAD) by kinases like Akt activates glucokinase to promote glycolysis and support the growth and proliferation of CSCs. 2 Dephosphorylated BAD directs cell death and dampens the metabolic pathways required for an elevated glycolysis to confer survival advantage of CSCs. 102
Metabolism-based therapy fires arrow at Achilles’ heel of CSCs
Through collective efforts dedicated to the development of new anticancer drugs targeting the metabolism of cancer cells, a variety of metabolism-based drugs have been tested in preclinical and clinical studies (Figure 3 and Table 2). Glucose deprivation by 3-bromopyruvate (3-BrPA) and analog 3-BrOP induced the in vitro elimination of CSCs and compromised in vivo tumorigenicity.17,103 2-DG could competitively inhibit glycolysis and block the generation of glucose 6-phosphate and inhibit HK2, and preferentially suppress the propagation of CSCs. 16 Pyruvate dehydrogenase kinase (PDK) inhibitor such as dichloroacetic acid (DCA) can induce the formation of PKM2/OCT4 complex and attenuate the transcriptional activity of OCT4, which modulates the apoptosis in glioma stem cells. 99 Vitamin C suppresses glycolysis 104 and miR-302/367 to compromise the reprogramming of breast CSCs through downregulation of TET1 gene. 105 It also inhibits stress-induced epinephrine-activated LDHA, and thereby decreases the lactate generation to dampen the USP28/MYC/SLUG pathway in CSCs. 101
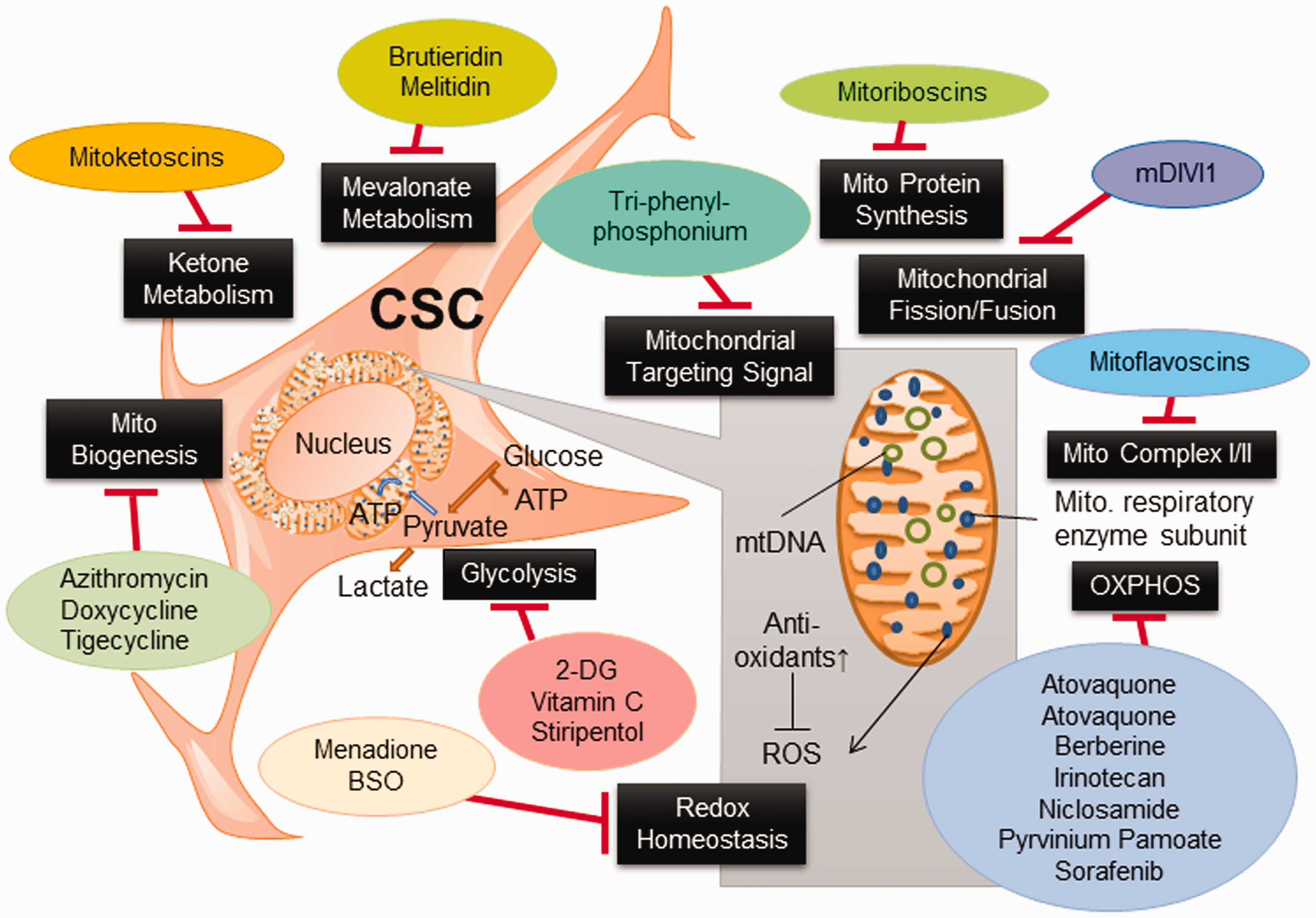
Metabolism-based therapy compromises CSC properties. Metabolism-based therapeutic strategies targeting pivotal metabolic pathways that can be exploited in preclinical and clinical settings. Specific metabolic inhibitors that can overwhelm stem cell properties may halt disease recurrence, attenuate CSC dissemination, and dampen the capacity of spawning metastasis of cancer cells. (A color version of this figure is available in the online journal.)
Metabolic inhibitors in different metabolic phenotypes.
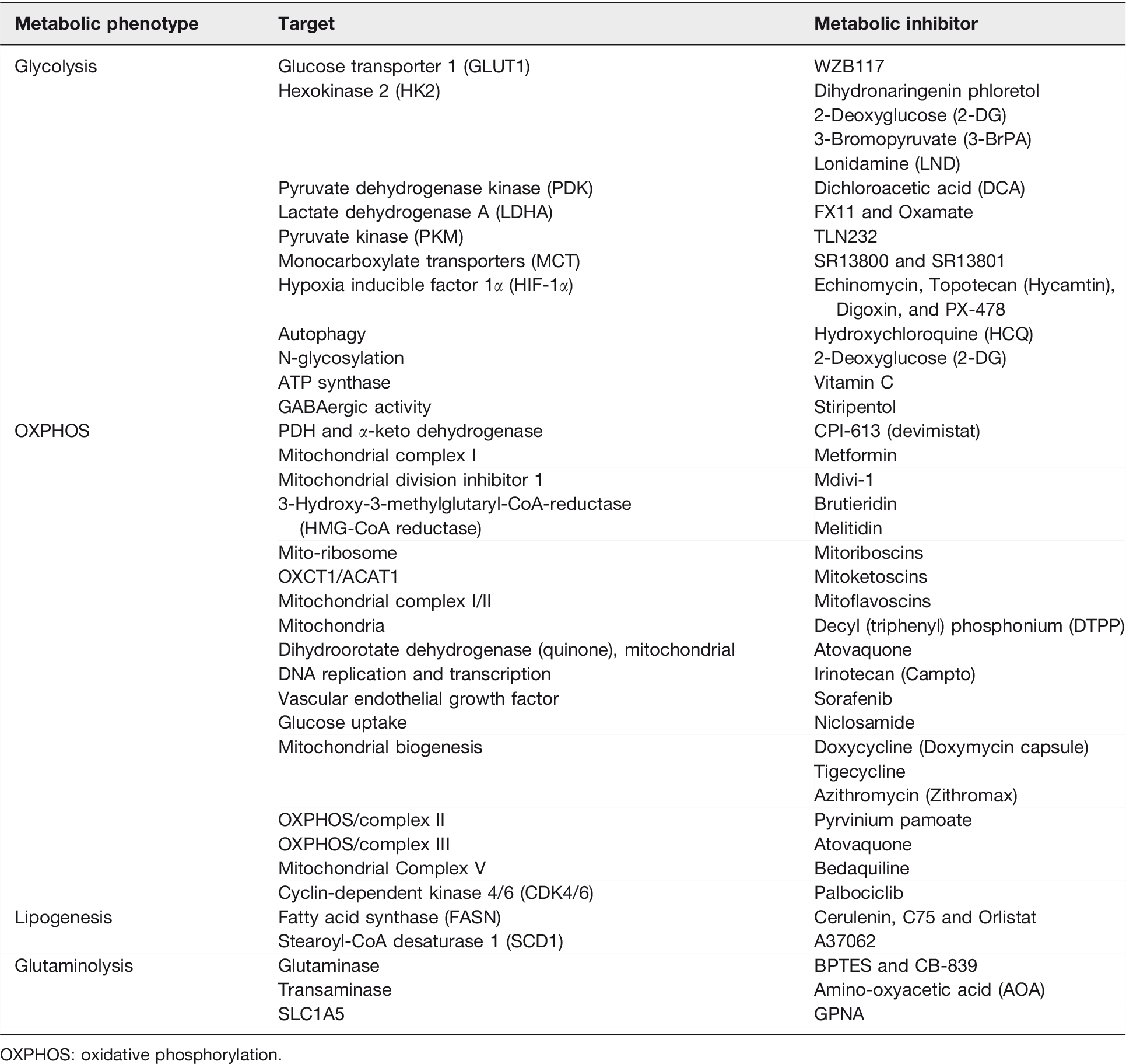
OXPHOS: oxidative phosphorylation.
As increased mitochondrial biogenesis and OXPHOS are observed in certain types of CSCs, mitochondrial inhibitors such as oligomycin, rotenone, antimycin A, metformin, or phenformin can lead to apoptosis of CSCs. 106 FAO inhibitor etomoxir can suppress CPT1 (carnitine palmitoyl-transferase 1) and re-sensitize CSCs to cytotoxic agents. 106 Based on the “endosymbiotic theory,” mitochondria in eukaryotic cells were initially derived from endocytosis of aerobic bacteria. Consequently, mitochondria still share some characteristics similar to bacteria, explaining the potential use of some FDA-approved antibiotics to target at mitochondria. 107 These antibiotics such as doxycycline and azithromycin were reported to impede the spherogenesis of CSCs through inhibition of mitochondrial function (Table 2). 107
Combination therapies encompassing conventional chemotherapies and chemo-sensitizing agents would be the most effective way to enhance the efficacy of CSCs-targeted therapy. 5-fluorouracil (5-FU)-refractory cells showed increased levels of PKM2 and acquisition of stemness in colon cancer. Co-treatment of 5-FU and metformin was found to dampen the respiratory chain Complex I activity, abolish spherogenesis of colon CSCs, and diminish the stemness markers. 108 Doxycycline resistant-CSCs are relatively more sensitive to metabolism-based drugs including OXPHOS inhibitors including atovaquone and irinotecan, glycolysis inhibitors such as vitamin C and stiripentol, as well as an autophagy inhibitor chloroquine (Table 2). 107 Emerging mitochondrial inhibitors “mitoketoscins” with special focus on ketone metabolism mimic the structure of coenzyme A, which functionally inhibit the activity of CSCs through binding to OXCT1 and ACAT1 catalytic sites within the binding sites of coenzyme A (Table 2). 109 In addition, diphenyleneiodonium chloride (DPI) halts mitochondrial respiration through the suppression of flavin-dependent enzymes, which constitute respiratory chain complexes I and II. Accordingly, DPI-induced chemoquiescence significantly reduces CSC subpopulation. 33 Moreover, a mitochondria-targeting compound, tri-phenyl phosphonium (TPP), can selectively annihilate both bulk tumor mass and CSCs but spare normal fibroblasts (Table 2). 110 TPP seems to be able to distinguish metabolically the mitochondria in normal cells from those in malignant cells because bulk tumor mass and CSCs likely have mitochondria with a higher Δψm. 111 Besides, targeting DRP1 protein by mDIVI1 compromised the mitochondrial fission–fusion cycles and mitochondrial function, cell migration, and CSC signaling (Table 2). 111 Naturally occurring mitochondrial inhibitors, Brutieridin and Melitidin, can suppress mevolonate metabolism to inhibit the propagation of CSCs (Table 2). 112 GLS1 inhibitors such as 968 or BPTES can attenuate CSC properties in hepatocellular carcinoma via increased ROS levels and impaired Wnt/β-catenin signaling (Table 2). 113 Glutaminase inhibitors, including Zaprinast (an anti-asthma drug) and BPTES, were able to effectively compromise the stemness and sensitize pancreatic CSCs to radiotherapy and enhance apoptosis in vitro and in vivo caused by redox imbalance. 48
Glycolysis and OXPHOS are two main metabolic engines in CSCs, which are not necessarily mutually exclusive or disjointed. It is worth mentioning that some CSCs manifest metabolic plasticity by switching to glycolytic phenotype when respiration pathway is blocked.28,114,115 In addition, the metabolic compensation with OXPHOS or other microenvironmental nutrient supply in CSCs may also render their resistance to inhibition of glycolysis. 13 Sequential or combinational treatment of two or more metabolic inhibitors would block the development of drug resistance and completely eradicate CSCs. For instance, “two metabolic hit strategy” proposes that the utilization of mitochondria-interfering agent like doxycycline drastically reverses the metabolism of CSCs toward an inflexible glycolytic pathway, rendering a second metabolic hit that completely halts the biochemical machinery of CSCs. 116 Metformin combined with either bromodomain and extraterminal motif (BET) or inhibitor JQ-1 for the treatment of pancreatic cancer or PI3K inhibitor for treating ovarian cancer can simultaneously halt OXPHOS and indirectly suppress glycolysis.20,117 Furthermore, CSCs do not become resistant to drugs upon administration of a mitochondrial ROS inducer menadione, implying that accumulated ROS levels to a toxic level may be an alternative strategy to increase the effectiveness of anticancer therapy to eradicate CSCs. 20 As a consequence, blocking glutathione synthesis by buthionine sulfoximine could disturb the redox homeostasis in CSCs, and subsequently decrease the clonogenicity and facilitating the radiotherapy responses of cancer patients.9,118
Because of the fact that some metabolic features of CSCs are common to those of normal stem cells, an accurate distinction between them is awaited. Once these metabolic differences can be identified, a novel therapy is able to be established to kill only CSCs but spare tissue stem cells. In such a scenario, simultaneous or sequential block of glycolytic flux and OXPHOS may be a new treatment to concurrently eliminate glycolytic and oxidative CSCs. These metabolic targets expose unrecognized Achilles’ heel in CSCs amendable for therapeutic intervention to prevent recurrence and thereby achieve long-term remission in cancer patients.
Future perspectives
CSCs may choose either glycolysis or OXPHOS as the major metabolic engine in response to environmental factors including tumor size, hypoxia, and the sequence of activation of oncogenes. The metabolic plasticity of the CSC subpopulation can be one of the major obstacles to eradicate CSCs. Therefore, a comprehensive portrait of the plasticity still awaits to be deciphered in CSCs. Instead of dwelling in a solitary place, CSCs reside in a diversified ecosystem encircled by extracellular matrix, immune cells, endothelial cells, and CAFs. Given that CSC–stromal interactions are quintessential for determining treatment response, limitations pertaining to the differences of tumor microenvironment can be minimized by devising better niche-targeting strategies. Thorough scrutiny of the singularities of mitostemness can provide clues to tackle CSCs by metabolic intervention. We envision that metabolic therapeutic interventions would eventually be practiced as an add-on to standard cytotoxic regimens, especially in halting cancer recurrence and metastasis to achieve more effective long-term disease remission. Lastly, discovering the metabolic distinction between CSCs and tissue stem cells is a prerequisite to develop new therapies that can aim at metabolically distinct CSCs but spare the normal tissue cells.
Footnotes
Authors’ contributions
YAS: conception and design, article writing. SCP: conception and design, first draft of article writing. IC and RYL: first draft of article writing. YHW: conception and design, article writing and revision, and administrative support.
DECLARATION OF CONFLICTING INTERESTS
The author(s) have declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
FUNDING
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This review has been prepared on the basis of the results obtained from research projects sponsored by grants (MOST104-2314-B-715–003-MY3, MOST104-2627-M-715–002, MOST104-2320-B-715–006-MY2, MOST105-2627-M-715–001, MOST106-2627-M-371–001, MOST106-2320-B-371–002, and MOST107-2320-B-371–002) from the Ministry of Science and Technology, Taiwan. The work in the laboratory of YA Shen was supported by a TMU grant 108–5403-003–112. The research work carried out in the laboratory of YH Wei that are included in this article have been supported by the above-mentioned grants and partly supported by intramural grants (Nos. 107-CCH-NPI-052 and No. 108-CCH-IST-149) from Changhua Christian Hospital, Changhua City, Taiwan.