
Abstract
The mechanism of aging is not yet fully understood. It has been recognized that there are age-dependent changes in the DNA methylation pattern of the whole genome. To date, there are several DNA methylation-based estimators of the chronological age. A majority of the estimators use the DNA methylation data from a single tissue type, such as blood. In 2013, for the first time, Steve Horvath reported the DNA methylation-based age estimator (353 CpGs were used) that could be applied to multiple tissues. A refined, more sensitive version that uses 391 CpGs was subsequently developed and validated in human cells, including fibroblasts. In this review, the age predicted by DNA methylation-based age estimator is referred to as DNAmAge, and the biological process controlling the progression of DNAmAge is referred to as the epigenetic aging in this minireview. The concepts of DNAmAge and epigenetic aging provide us opportunities to discover previously unrecognized biological events controlling aging. In this article, we discuss the frequently asked questions regarding DNAmAge and the epigenetic aging by introducing recent studies of ours and others. We focus on addressing the following questions: (1) Is there any synchronization of DNAmAge between cells in a human body?, (2) Can we use in vitro (cell culture) systems to study the epigenetic aging?, (3) Is there an age limit of DNAmAge?, and (4) Is it possible to change the speed and direction of the epigenetic aging? We describe our current understandings to these questions and outline potential future directions.
Impact statement
Aging is associated with DNA methylation (DNAm) changes. Recent advancement of the whole-genome DNAm analysis technology allowed scientists to develop DNAm-based age estimators. A majority of these estimators use DNAm data from a single tissue type such as blood. In 2013, a multi-tissue age estimator using DNAm pattern of 353 CpGs was developed by Steve Horvath. This estimator was named “epigenetic clock”, and the improved version using DNAm pattern of 391 CpGs was developed in 2018. The estimated age by epigenetic clock is named DNAmAge. DNAmAge can be used as a biomarker of aging predicting the risk of age-associated diseases and mortality. Although the DNAm-based age estimators were developed, the mechanism of epigenetic aging is still enigmatic. The biological significance of epigenetic aging is not well understood, either. This minireview discusses the current understanding of the mechanism of epigenetic aging and the future direction of aging research.
Introduction
A fundamental question regarding aging is whether there are time-dependent molecular (or chemical) changes controlling aging. One attractive explanation is the telomere shortening after every cell division (reviewed in Turner et al. 1 ). Telomere length determines the limit of cell divisions, and this limit may be one of the determinants of the lifespan of animals. 1 The telomere theory is attractive. For example, the risk of death certainly increases by age if the blood cell supply decreases due to the telomere shortening of the hematopoietic stem cells (HSCs) that need to divide to produce blood cells. However, the telomere length cannot simply explain how the lifespan is controlled. It is known that mice have much longer telomeres than humans, though the lifespan of these animals is considerably shorter than that of humans. 2 In fact, the first generation of the telomerase knock-out mice is viable, and the severe age-associated diseases develop after multiple generations of the mutant mice. 3 These studies suggest that there are multiple factors determining the lifespan of mammals in addition to the telomere length.
Age-dependent changes of DNA methylation (DNAm) have long been described in both mice and humans, as well as other organisms.4–6 These age-dependent DNAm changes were found in hematopoietic cells, fibroblasts, neurons, and other cell types. Because of the convenience of the sample collection, the analysis of age-dependent DNAm has been very well studied in blood cells (for example, see literature7–9). There are only a handful of enzymes directly controlling DNA methylations events including DNA methyl-transferases, DNMT3L, DNMT1, 3a, and 3 b, and DNA “de-methylating” enzymes, the hydroxylases ten-eleven translocation (TET) (TETs: TET1, 2, and 3),4,10,11 but their contribution to age-dependent epigenetic changes has not been thoroughly elucidated. Several hypotheses have attempted to address this question such as the scars after DNA damage repair and the cell division, accumulation of entropy of DNAm pattern among others.4–6
Epigenetic clock: Prediction of the age by DNAm analysis
Based on the analysis of age-dependent DNAm change, several programs have been developed predicting the age of the cells by their DNAm pattern.4–6 The majority of these programs uses DNAm data from a specific set of cell type; for example, the blood cells.7–9 In 2013, Horvath 12 reported a new age estimator that can be applied to multiple human tissues. This multi-tissue age estimator is referred as to the epigenetic clock. The epigenetic clock uses DNA methylation patterns of the specific 353 CpG sites. Horvath recently developed a refined epigenetic clock that predicts chronological age significantly more accurately, especially when it is applied to skin cells and cultured fibroblasts. 13 In this new epigenetic clock, 391 CpGs were selected to predict the chronological age, 13 and it is considered the most accurate age estimator based on DNAm at present. 14 In the current manuscript, the predicted age by the epigenetic clock is referred to as the DNAmAge, and the biological process controlling the progression of DNAmAge is referred to as the epigenetic aging. 4 Horvath et al.15–20 published studies showing that the acceleration of DNAmAge progression (i.e., faster epigenetic aging than the actual chronological age) is associated with a higher incidence of life-limiting diseases and mortality. These studies support the usefulness of the DNAmAge as a biomarker of aging predicting the risk of age-associated diseases.
Frequently asked questions of DNAmAge and epigenetic clock
FAQ 1: Is there a synchronizing mechanism of DNAmAge in the human body?
The interesting fact is that DNAmAge predicts the chronological age of various types of cells that have different histories of differentiation and cell division. This fact raises a question; Is there any synchronization mechanism of the epigenetic clock in a human body? We addressed this question by analyzing the DNAmAge of blood cells of leukemia survivors who received hematopoietic stem cell (HSC) transfer from donors who were more than 10 years younger or older than the recipient. 21 As we discuss in-depth later, our data suggest that the blood cells have their own intrinsic epigenetic aging mechanism.
F: Is there an age limit for the DNAmAge?
Presently, the verified longest-lived human is 122 years old. 22 If the DNAmAge accurately reflects the age of human tissues, can we use the concept of DNAmAge to predict the possible maximum lifespan (age limit) of humans? At present, we do not have the answer. This question will be discussed by focusing on the DNAmAge analysis of cultured cells as well as cancer cells in this article.
F: Can we use the cell culture system to study the mechanism and the biological significance of DNAmAge?
The short answer is yes. There are reports demonstrating the usefulness of cell culture systems to investigate epigenetic aging.13,23–26 We will discuss the pros and cons of cell culture models in this article. Of noted, the calculation of DNAmAge uses the DNAm rate of 353 or 391 CpGs based on a genome-wide DNAm analysis of DNA samples (e.g. one microgram DNA) extracted from approximately one million cells.12,13 Therefore, it is safe to claim that DNAmAge is the aging biomarker of the cell population but not of every single cell. 4 An old DNAmAge of one DNA sample from one tissue (for example, one million cells) means that the ratio of cells with age-dependent DNAm change is high. This condition does not mean that all cells have “old DNAm pattern”.
F: Can we reverse or slow the progression of DNAmAge?
The short answer is yes, at least in experimental conditions. In other instances, the rejuvenation of DNAmAge is commonly observed in cancer cells in which normal epigenetic aging system is destroyed. 12 The most drastic approach would be to reprogram the DNAm pattern by generating induced-pluripotent stem (iPS) cells whose DNAmAge was reset to 0 years old. 12 However, this reprogramming method is not feasible in humans. To date, attempts to discover strategies to reverse or delay DNAmAge progression are being made by numerous researchers. For example, rapamycin treatment has been shown to slow DNAmAge progression in vitro. 27 Very recently, it has been shown that DNAmAge was rejuvenated in human muscle stem cells by the transient expression of the reprogramming factors used for iPS production. 28 Moreover, we reported that hypoxia condition 24 and G-CSF treatment 21 have effects of slowing or slightly reversing DNAmAge progression, respectively. We will discuss these observations in-depth in this article.
Hematopoietic cells have an intrinsic epigenetic aging mechanism (Figure 1)
An interesting fact is that cells from different tissues from the same individual exhibit similar DNAmAge. 13 For example, lymphocytes, skin fibroblasts, hepatocytes, and cheek epithelial cells (CECs) displayed the DNAmAge that matched the host’s chronological age. It is not yet known why different cell types show the same DNAmAge. In our recent study, we examined the DNAmAge of blood in hematopoietic stem cell transplant (HSCT) recipients. We specifically selected donor–recipient pairs with more than 10 years of age difference (up to 50 years age difference). 21 We found that the DNAmAge of HSCT recipients matched the chronological age of the donor even 17 years after HSCT. 21 We assessed recipients who received HSCT from younger or older donors. In both cases, DNAmAge of blood matched the donor’s age but did not correlate with the recipient patient’s age. These results suggest that the progression of DNAmAge in hematopoietic cells is a cell-intrinsic mechanism, and rejuvenation or accelerated progression of DNAmAge did not occur by the bone marrow niche or other host cells. Stolzel et al. 29 also reported a similar finding by investigating the DNAmAge of the leukemia survivors who received HSCT. Although another type of DNAm-based age estimator was used, Wagner’s group examined the speed of the epigenetic aging of human blood cells after the transfer of these cells to the irradiated humanized mice. 30 They found that the epigenetic age of circulating human blood cells was not influenced by the mouse body. 30 Recently, it was reported that the progression of DNAmAge was observed in cultured human retinal tissue and organoids. 26 These results suggest that the epigenetic clock can function in each tissue without the contribution from circulating blood cells. Although these results suggest that the hematopoietic system (i.e. blood cells) as well as the retinal tissue (and probably other tissues) have their own intrinsic epigenetic aging mechanism, the possibility remains that circulating blood cells have a certain influence on the epigenetic aging of non-blood cells. We are investigating this latter possibility and hope to report our findings in the near future.
Granulocyte-colony stimulating factor and DNAmAge
For patients undergoing HSCT, there are three primary strategies to obtain stem cells (HSCs) 31 : (1) directly from the bone marrow (BM), (2) from peripheral blood using granulocyte-colony stimulating factor (G-CSF) treatment to mobilize HSCs, and (3) HSCs from umbilical cord blood. Given that most cord blood recipients are children and the restrictions involved when accruing children for tissue collection, our studies were conducted solely in adult leukemia survivors treated with peripheral blood stem cell transplant or bone marrow-derived aspirate. 21 We determined that although the DNAmAge of HSCT recipients closely correlated with donor age, there was a significant difference in DNAmAge between the two sources of HSCs. Blood originating from G-CSF-treated donors showed a DNAmAge approximately five years younger than the chronological age of the donor. 21 However, this “rejuvenating effect” was not observed in recipients that had received BM-derived HSC. 21 Recent studies showed that G-CSF treatment has beneficial effects against age-associated health problems such as neurodegeneration32–34 and cardiovascular dysfunction.35–37 However, studies have not addressed if the effects of G-CSF on overall host health are due to an anti-aging property. Future studies are clearly warranted to determine if G-CSF modifies the epigenetic age of blood and host tissues in blood cancer survivors.
G-CSF is a cytokine regulating hematopoietic cell proliferation, differentiation, and function. 38 It plays a vital role in the production of neutrophils and stimulates granulocyte colony formation from hematopoietic progenitor cells. 38 The mechanism of HSC mobilization is not through the direct actions of G-CSF on bone marrow HSCs. Rather, it is thought that G-CSF activates metalloproteases of BM niche cells, which in turn cleave vascular cell adhesion protein 1 (VCAM-1) 39 and promote the release of norepinephrine from the sympathetic nervous system, 38 leading to the release of bone marrow stem cells into the peripheral blood. Since G-CSF may not directly act on HSCs, we speculate that its effect on DNAmAge may be a consequence of (1) enhanced cell release from the bone marrow into the circulation, and (2) the transplantation process. Another possibility is that the G-CSF indirectlty influences the DNAmAge of HSCs through other G-CSF target cells. There are many remaining experiments to be performed to understand the effects of G-CSF on the DNAmAge of blood cells.
Abnormal DNAmAge progression in leukemia cells
It has been known that epigenetic aging is not operative in cancer cells. 4 Cancer cells show abnormally old or young DNAmAge that does not match the chronological age of the cancer patient. 12 In the case of acute myeloid leukemia (AML) cells, we also observed that the DNAmAge is significantly more advanced or decreased in comparison with the chronological age of the patient(e.g., AML cells from 50-year-old AML patients displayed a DNAmAge of 248 years). 21 In AML, it is known that the enzymes regulating DNA-methylation are often mutated or silenced. 11 Therefore, abnormal DNAmAge is likely due to these mutations in AML cells. To our surprise, however, we also uncovered that abnormal DNAmAge of normal blood cells preceded clinical relapse of leukemia in patients. We discuss this intriguing observation in the next paragraph.
Abnormal progression of DNAmAge of normal blood cells precedes relapse of leukemia
In the leukemia survivors who did not experience disease relapse, the DNAmAge of blood cells from HSC recipients (i.e. leukemia survivors) matched the chronological age of the donors (r = 0.89, n = 24, P < 0.0001). 21 Leukemia patients that eventually relapsed but at the time had no evidence of active disease (donor chimerism was >95%), the DNAmAge of peripheral blood cells showed moderate correlation with donor age (r = 0.61, n=12, P = 0.04) which is a weaker correlation in the case of the patients with no relapse. 21 These results suggest that DNAmAge can be used to predict relapse risk in blood cancer patients. 21 For example, the abnormal acceleration or rejuvenation of the DNAge of the blood of HSCT recipients predicts the relapse of AML several months before the relapse. However, the precise mechanisms by which leukemia cells impact the DNAmAge of healthy bone marrow cells are not known. We speculate that changes in DNAmAge of blood cells are induced by an immunologic response to residual cancer cells. Therefore, further studies are warranted to determine the clinical utility of DNAmAge in predicting the risk of relapse at considerably earlier time points than currently available methods, including donor chimerism or the detection of cancer cells by flow cytometry or molecular techniques.
Is there any age limit for DNAmAge?
DNAmAge is calculated by the sum of the DNAm changes of 353 or 391 CpGs.12,13 Therefore, technically speaking, DNAmAge can increase as much as the calculation allows. DNAmAge can even be negative when DNAm of embryonic cells was measured. 26 As far as we know, the maximum (i.e. oldest) DNAmAge identified in cells or tissues to date is “248 years-old” which was reported in our recent study examining the DNAmAge among HSCT recipients. 21 This DNAmAge was obtained from blood samples of a patient with relapsed AML. Therefore, this DNAmAge is due to the presence of cancer cells that have abnormal DNAm patterns. In this patient, AML cells did not show aneuploidy, and the chromosome number was normal corroborating that the abnormal DNAmAge was not due to abnormally increased number of chromosomes.
Cancer cells evade Hayflick limits, and keep dividing with no check point inhibition. 40 The mechanisms of immortalization (unlimited cell division) of cancer cells include the telomere maintenance by an increase in telomerase expression and the suppression of cellular senescence.41,42 Many well-established cancer cell lines show a DNAmAge in the range of 10–140 years. 12 For example, MDA-BB 231 and MDA-MB-468 are commonly used breast cancer cell lines that originated from a 51-year-old woman. The DNAmAge of these cell lines is 138.18 years (MDA-MB231) and 11.14 years (MDA-MB-468). 12 Many cancer cell lines have aneuploidy. In fact, both MDA-MB-231 and MDA-MB-468 have near-triploid chromosomes (modal number = 64, MDA-MB-231: range 52 to 68, MDA-MB-468: range 60–67). Interestingly, the abnormally increased number of chromosomes does not necessarily increase the DNAmAge even though some of the DNAmAge-defining-CpGs are duplicated or triplicated. At present, it is not yet determined whether DNAmAge of cancer cells can advance beyond “248 years-old” by the additional several years of cell cultures. DNAmAge may increase continuously or it may not be able to advance beyond a certain level due to an undisclosed ceiling factor.
Elovl2 and the DNAmAge
Among 353 CpGs and 391 CpGs defining DNAmAge, there are several sites that have been known to show the age-dependent hypermethylation. The CpG in the Elovl2 gene promoter region is an example.12,13 The age-dependent hypermethylation of the Elovl2 gene has been recognized as an epigenetic marker of aging,7,8 and has been confirmed across multiple mammalian species (mouse, dog, monkey, and human). 43 The name of Elovl2 comes from "elongation of very-long-chain fatty acids-like 2 (ELOVL2)" gene which encodes a transmembrane enzyme involved in elongation of long-chain polyunsaturated fatty acids.44,45 This enzymatic activity of Elovl2 contributes the synthesis of docosahexaenoic acid (DHA) which is one of the main polyunsaturated fatty acids in the human nervous system.44,45 Recently, it has been reported that the age-dependent decrease of Elovl2 expression may cause age-associated macular degeneration (AMD)-like retinal degeneration in the mouse. 46 The Elovl2 CpG site may have an evolutionarily conserved role in regulating the onset of age-associated physiological changes in mammals.25,43 The age-dependent decrease of ELOVL2 expression may result in the decrease of the utilization of DHA and other polyunsaturated fatty acids, and this decrease may cause degenerative changes of the aged tissue.
SH3BP5 expression is elevated in the skeletal muscle of aged individuals (Figure 2)
The CpGs in SH3BP5 (SH3 Binding Protein 5) gene are part of the CpGs defining DNAmAge, and show age-dependent hypomethylation.12,13 Interestingly, the expression levels of SHBP5 mRNA in skeletal muscle increase with age in both human and mouse, and this increase is attenuated by calorie restriction in mice (Figure 2). To our knowledge, there are no reports about the role of SH3BP5 in age-dependent degenerative diseases. Although further studies are needed to reach this conclusion, the age-dependent hypomethylation of CpGs in the SH3BP5 gene may be causally related to increased gene expression of SH3BP5. Previous studies suggest that SH3BP5 has a pro-apoptotic activity inducing mitochondria-dependent cell death pathway.47–50Age-dependent elevation of SH3BP5 expression may facilitate age-dependent sarcopenia by inducing apoptosis of muscle cells.
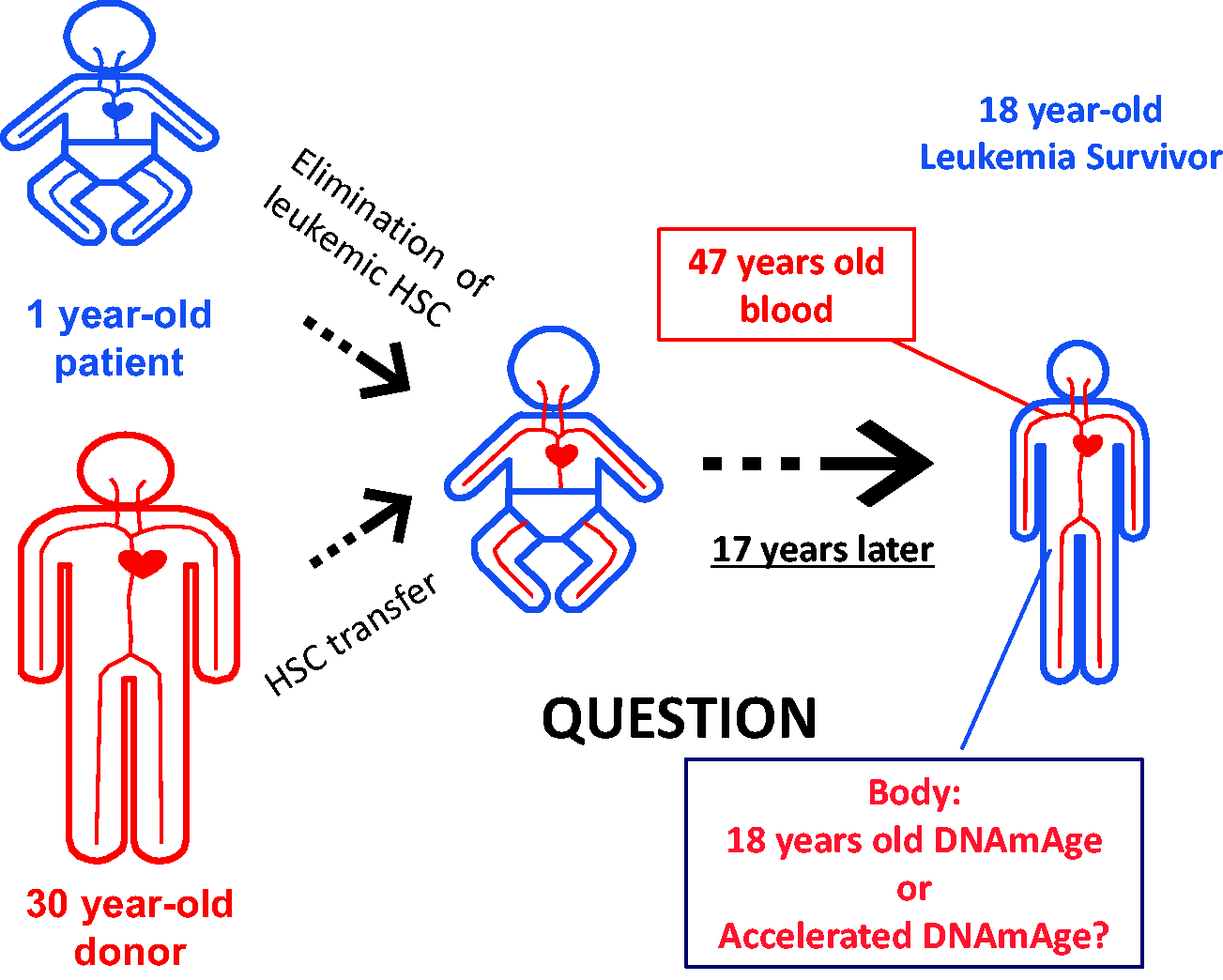
Hematopoietic Stem Cell Transfer (HSC) from an old donor to a young recipient. The DNAmAge of transplanted blood cells progressed in parallel with blood cells of the donor for over 17 years. The remaining question is whether these donor blood cells influence the DNAmAge of non-hematopoietic cells in the recipient individual. (A color version of this figure is available in the online journal.)
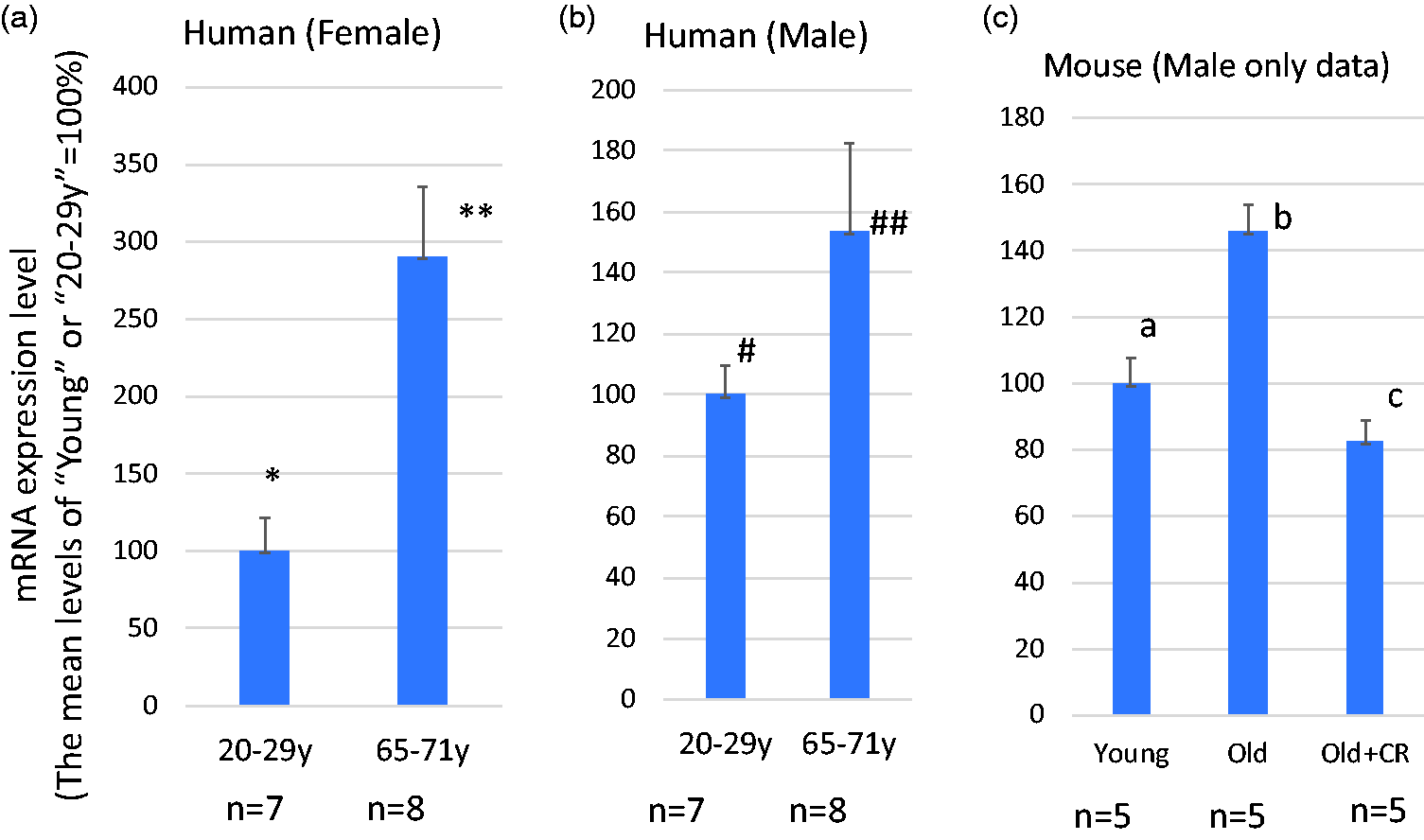
The levels of SH3BP5 gene expression increase by aging. (a and b) SH3BP5 mRNA expression in young and old females (a) and males (b). The increase of SH3BP5 expression is statistically significant in the female group (* vs.**P < 0.005) with a trend towards significance between older and young males (# vs. ##P = 0.129). These data were obtained from the Gene Ontology Database (National Institute of Health (NIH)), ID=GDS472:201810_s_at and ID=GDS472:201811_x_at). (c) The effect of calorie restriction on SH3BP5 mRNA expression in mouse skeletal muscle. Data were obtained from C57B6 male mice, ages 5 months (young) and 25 months (old). Mice were fed a normal or calorie restricted (CR) diet (details are described in BMC Genomics 2007 Mar 23;8:80. PMID: 17381838). These data were obtained from the Gene Ontology Database (National Institute of Health (NIH)), ID= GDS2612/1421923_at). a vs. b: P < 0.003, b vs. c: P < 0.0003, a vs. c: P = 0.269. (A color version of this figure is available in the online journal.)
Cell culture model
To date, there are several reports describing the use of cell culture systems to investigate the mechanisms controlling DNAmAge progression.13,23–26 Importantly, it has been shown that DNAmAge is not functionally correlated with telomere shortening and cellular senescence that are known to be the hallmarks events of aging. 51 For example, studies showed that (1) DNAmAge progression did not stop even when cells were immortalized by hTERT expression, and that (2) cellular senescence occurs at any DNAmAge by oncogene overexpression or radiation-induced DNA damage.23,51 Recently, we reported that DNAmAge progression can be monitored over six weeks of fibroblast cell culture, and that the speed of DNAmAge progression was affected by oxygen level, the chronological age of the cell, and tumor suppressor status, 24 as discussed in the next paragraph.
DNAmAge progression of cultured fibroblast is faster than the chronological time progression
In our recent studies, we showed that the speed of DNAmAge progression in cell culture is five times faster than the chronological time progression when cells were cultured in the 5% CO2 incubator (with 95% air). 24 Reports from other groups also showed that DNAmAge progression of fibroblasts is accelerated in cell culture. 25 Interestingly, in the case of the cultured retinal organoid, the speed of DNAmAge progression matched the chronological time progression. 26 At present, we do not have an evidence-based explanation of why accelerated DNAmAge progression was observed in the fibroblast culture, but not in the retinal organoid culture. One possibility is the difference in cell proliferation activities. The fibroblasts cell culture system allows cells to continuously divide by performing cell passages, whereas the organoid culture system has regulated cell division activity similar to in vivo conditions.
Hypoxia slows DNAmAge progression in dividing cells
Conventional cell culture is performed in a CO2 incubator that maintains 5% CO2 and 95% air. Air contains 21% oxygen, and this is referred to as the normoxic state. In the human body, oxygen levels are maintained lower than atomospheric air, for example, the levels are 2–3% in the brain, liver, and kidney; or 1–2% in the hematopoietic stem cell niche.52,53 To determine the effects of oxygen on DNAmAge, we cultured fibroblasts for six weeks in a 5% CO2 incubator under hypoxic (1% O2) conditions. 24 Interestingly, the progression of DNAmAge was significantly attenuated in hypoxia by approximately 40% compared to normoxia. 24 It is known that the dividing speed slows in hypoxia in comparison with normoxia. 54 On the other hand, hypoxia suppresses the onset of cellular senescence induced by oxidative stress. 54 After six weeks culture (eight passages), the total cell doubling times in hypoxia was actually higher than that in normoxia, whereas the DNAmAge progression was slower in hypoxia. 24 These results indicate that hypoxia slows the DNAmAge progression even though it increased the total number of cell doubling time. 24 To be noted, the speed of DNAmAge progression of cultured fibroblast in hypoxia is still faster than the chronological time speed. 24 As we discussed, the continuous cell division in vitro may be a reason for this acceleration. In addition, high glucose levels in the culture media may be another factor for the acceleration in DNAmAge progression. 25
Hypoxia inhibits the ubiquitin-proteasome-dependent degradation of hypoxia-induced factor 1α (HIF1α) protein, and therefore HIF1α levels are maintained high in hypoxia. 54 HIF1α is a transcription factor controlling the expression of various genes including enzymes controlling DNAm. A previous report showed that HIF1α decreases the expression of DNA methyltransferases (DNMT1, 3 A, and 3B), whereas it increases DNA de-methylating enzymes (TET-2 and -3). 55 Therefore, hypoxia may slow the progression of DNAmAge by altering the balance of DNAm modifying enzymes though HIF1α. If there are specific clusters of DNAmAge-defining CpGs influenced by hypoxia, these CpGs will be useful tools to investigate the mechanisms by which oxygen tension influences epigenetic aging.
Immortalization and DNAmAge
In human cells, hTERT overexpression and p53 inactivation are the most commonly used methods to generate an immortalized cell line. 56 It has been shown that hTERT-mediated immortalization does not “stop” the progression of DNAmAge in cell culture.51,57 This is a very intriguing finding since immortalized cells continued to age at the epigenetic level. However, it is not yet tested how far the DNAmAge progression goes by hTERT-mediated immortalization. hTERT-mediated immortalization does not stop DNAmAge progression after several months of cell culture experiments, but whether there is a peak DNAmAge that can be achieved in cell culture, is not presently known.
We also examined the effects of SV40 Large T expression that is another frequently used method to induce immortalization of cultured cells. SV40 Large T is known to prevent cellular senescence by mainly inactivating p53. p53 is a tumor suppressor that induces the expression of proteins essential for cellular senescence and apoptosis which constitute two major cellular responses preventing cancer development. 42 SV40 Large T-mediated immortalization resulted in the randomness of DNAmAge changes during long-term cell culture. The decrease (rejuvenation) in DNAmAge was observed after six (6) weeks of culture in some dishes, whereas increase of DNAmAge occurred in other dishes. 24 As we discussed earlier, DNAmAge of cancer cells does not match with the age of the originating patients, and DNAmAge progression in cancer cells is random. These observations suggest that p53 and tumor suppressor genes play essential roles in maintaining the epigenetic aging mechanism intact so that it progresses in one direction along with the time progression. At present, we do not have clear answers about how tumor suppressors control the direction of the epigenetic aging. The p53 protein may become a useful tool to investigate how one-directional progression of DNAmAge is controlled, and this type of mechanistic study may open a new research field uncovering the mechanism of epigenetic aging.
Summary
In this article, we discuss the biological significance of DNAmAge and the molecular mechanisms of the epigenetic aging. The usefulness of DNAmAge as an aging biomarker is promising. At the same time, the concept of the epigenetic aging provides various opportunities for basic scientists to investigate the mechanisms of aging. The DNAmAge of the embryonic stem (ES) cells or iPS cells is 0 years. 12 Interestingly, the induction of differentiation starts the progression of DNAmAge in cell culture. 26 Investigations into the mechanisms that initiate and propagate epigenetic aging should include regulation of DNAm modifying enzymes (DNMTs and TETs) as well as other indirect mechanisms (e.g. chromosome structure or histone modification) controlling access of these enzymes to CpGs defining DNAmAge. DNAmAge provides a new paradigm of epigenetic control of cellular function. In addition to the differentiation status, we now know that the DNAm pattern can tell us the age of the tissue. As we discussed here, it is not yet clear how DNAmAge is correlated with cellular function. Many important questions remain to be investigated (Figure 3).
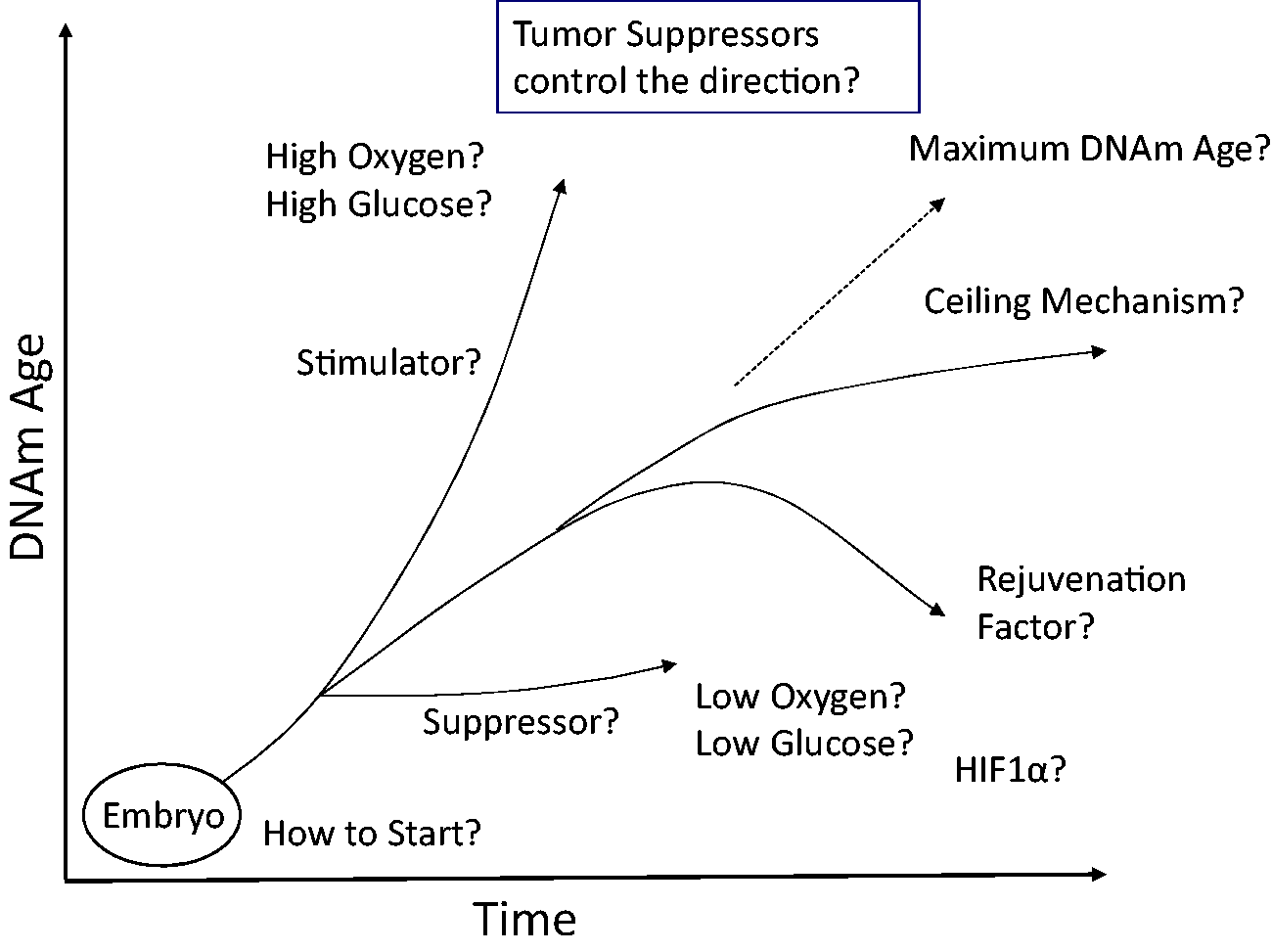
Remaining key questions. This figure summarizes the remaining questions regarding the epigenetic clock and DNAmAge.
Footnotes
Authors’ contributions
The first draft of this manuscript was prepared by SM. All co-authors participated in writing by improving the first draft (correcting errors, adding necessary information, and editing sentences). After all the authors contributed to writing, the final draft was prepared for submission.
DECLARATION OF CONFLICTING INTERESTS
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this manuscript.
FUNDING
This study was supported by National Institute of Health 1U34AG051425‐01 (University of California, Los Angeles) and P30CA043703 (Hematopoietic Biorepository and Cellular Therapy Facility of the Case Comprehensive Cancer Center); The South‐Eastern Norway Health Authority, Early Career Grant 2016058; The South‐Eastern Norway Regional Health Authority Project Grant 2017092; The Norwegian Cancer Association Grant 190278 and Raagholtstiftelsen.