
Abstract
Autophagy plays a crucial role in cellular development and differentiation as well as in the maintenance of homeostasis in healthy cells. Autophagy is well documented in neurodegenerative disorders, aging, and infectious diseases. However, recognizing its significance in cancer has always been challenging due to its tumor-promoting and suppressive attributes. Various modulators targeting key components of autophagy machinery directly or indirectly have been developed over the years, and have shown promising results in preclinical models. Some of these compounds are even being tested in clinical trials for safety and efficacy. A detailed review of strategies used to target autophagy in cancer is presented including our opinion on developing better therapies and outstanding issues.
Impact statement
A growing body of evidence has implicated the involvement of autophagy in several diseases, including cancer, over the last few years. This catabolic process is also essential for maintaining cell survival and homeostasis in normal cells. Although drugs targeting intermediates in autophagy signaling pathway are being developed and tested at a preclinical level as well as in the clinic, we still have a long way to go in terms of identifying an effective treatment strategy, considering the complex role of autophagy in cancer. This article sheds some light on currently implemented strategies to exploit the dependence of cancer cells on autophagy, and challenges in the field. We hope that the outstanding questions discussed in this article will encourage scientists to look at unexplored links between autophagy and other signaling pathways, which may lead to the discovery of novel, highly specific interventions targeting the process.
Introduction
Cancer is one of the leading causes of death all over the world and is the second leading cause of death in the United States. In 2020, it was projected that 1,806,590 new cancer cases and 606,520 cancer deaths could occur in the United States.1,2 Abnormal proliferation of cells in the body leads to the formation of a tumor. It can affect any tissue or organ. Normally, our immune system will recognize and eliminate it but sometimes, cancer cells evade the immune surveillance to become immortal by acquiring many features such as genome heterogeneity, migration and infiltration to local as well as distant tissues (metastasis) through blood or lymph nodes,3,4 initiation of angiogenesis to acquire nutrients, and resistance to programmed cell death.5,6 They hijack signaling pathways relating to normal cell growth to become self-sufficient in growth signals (growth factors, extracellular matrix components, adhesion molecules), and are insensitive to anti-proliferative signaling (cell cycle and other related signaling).1,7 In addition, cancer cells avoid apoptosis by regulating cell survival signaling. 8
For two decades, researchers have been focused on understanding the molecular mechanism of cancer cell survival and fundamentals of the uncontrolled growth. Researchers are fascinated by how cancer cells can become insensitive to cell death triggered by internal and external stimuli. Autophagy represents an internal stimulus in which a catabolic program brings about the degradation of cellular components through lysosomes. Autophagy is a highly coordinated “self-eating” process employed by cells to eliminate unwanted or damaged organelles. Autophagy is essential during normal development to maintain cellular homeostasis, 9 and regulate cell division and differentiation. 10 Alterations in autophagy have been reported in diseases such as neurodegenerative disorders, infectious diseases, liver diseases, diabetes, and aging. 11 In cancer, autophagy acts as a tumor promoter as well as suppressor.12,13 Due to its conflicting role in cancer, autophagy has become a popular therapeutic target in recent years. This review focuses on the overview of autophagy, its paradoxical role in cancer, and interventions targeting autophagy process.
Overview of autophagy
Autophagy is characterized by four phases—initiation, nucleation of autophagosomes, maturation, and degradation (Figure 1)–which are regulated by 5ʹ adenosine monophosphate-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) pathways.14,15 Under low-nutrient conditions, AMPK is activated and mTOR is inhibited. This results in ULK complex activation, which then phosphorylates Beclin-1 and activates VPS34, leading to nucleation and phagophore formation. 16 Class III phosphatidylinositol 3-kinase (PI3K) complex plays a key role in this process. Autophagy-related (ATG) proteins enable lipidation and subsequent incorporation of LC3 protein into the phagophore membrane. 17 This leads to the formation of autophagosome, which upon fusion with the lysosome, brings about degradation of its contents. When Beclin-1 interacts with B-cell lymphoma-2 (BCL-2), the nucleation process is inhibited, thereby inhibiting autophagy. Various factors such as nutrient deprivation, stress, hypoxia, elevated levels of reactive oxygen species (ROS), and anti-cancer drug treatments can trigger autophagy in cells. 18
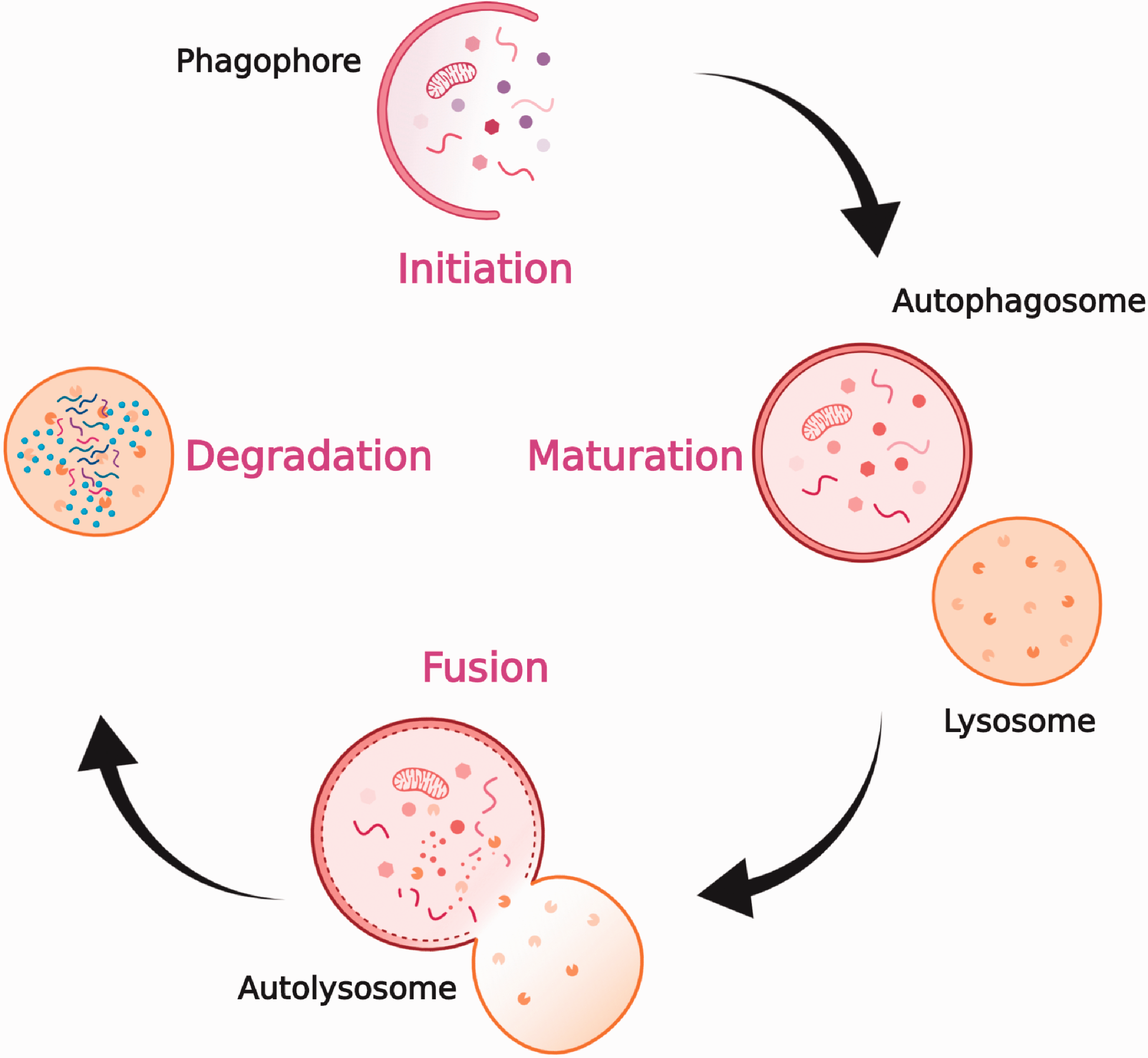
Overview of autophagy. Upon induction of autophagy, phagophore is formed, cytoplasmic materials are seized by a double-membraned structure, called the autophagosome. These autophagosomes fuse with lysosomes to become autolysosomes, where the seized cargo is degraded and recycled. Figure created with BioRender. (A color version of this figure is available in the online journal.)
A paradoxical role of autophagy in cancer
Some discrepancies exist in terms of the function of autophagy as tumor suppressor or promoter. Its role depends on the extent of cancer spread. Studies have shown that autophagy suppresses the commencement and progression of tumors. 19 On the other hand, autophagy is also known to play an important role in the survival pathway in response to stress. 20 Previous reports have shown that well-developed tumors require autophagy for cell survival. 18
Tumor suppression
Autophagy functions as a piece of key machinery for tumor suppression at the basal level via degradation of damaged cellular organelles and proteins, and maintenance of cellular homeostasis. 21 Monoallelic deletions of BECN1, a key autophagy gene encoding Beclin-1 protein, were found in prostate cancers, breast cancers, ovarian cancers, and hepatocellular carcinoma (HCC).22,23 Beclin-1 is known as a classical tumor suppressor because only a single copy is adequate to maintain its function in normal cells. BECN1 mutation causes impairment or loss of Beclin-1, inducing the formation of neoplasm. 24
ATG proteins are key players in the regulation of autophagy. Gastric and colorectal cancer patients were reported to have frameshift mutations in ATG2B, ATG5, ATG9B, and ATG12 genes, indicating defective autophagy in cells 25 The point mutations and deletions of ATG5 gene have been reported in gastrointestinal and other cancers. 19 On the other hand, the inhibition of tumor growth was observed following the expression of ATG16L1 in cancerous epithelial cells, revealing the compartment-specific role of autophagy. 26 In addition, autophagy suppresses tumor formation by preventing the accumulation of p62/SQSTM1, which is an adapter protein and a substrate of autophagy. Abnormal p62 levels have been observed in many cancers. 19 Moreover, there are indications that tissue necrosis, genomic instability, and chronic inflammation occurring as a result of deregulated autophagy, can elevate cancer risk by causing stress and increasing the occurrence of mutations. 27 These findings support the notion that intact autophagy process is essential for preventing tumorigenesis.
Hypoxic or nutrient-deprived conditions activate autophagy, enabling cell survival in cancer. On the other hand, apoptosis inhibits tumor cell survival. This leads to the crosstalk between various apoptosis and autophagy proteins. For example, autophagy protein ATG12 inhibits myeloid cell leukemia sequence 1 (MCL-1), a pro-survival member of BCL-2, to bring about mitochondrial apoptosis. 28
Tumor suppressors such as PDCD4 (programmed cell death protein 4) and p53 use autophagy inhibition as a mechanism underlying their tumor suppressive action. PDCD4 inhibits the expression of ATG5 and the formation of an ATG12-ATG5 complex, eventually inhibiting autophagy and tumor formation. 29 Likewise, several studies have shown that cytoplasmic p53 inhibits autophagy independent of its role as a transcriptional factor.30–32
Autophagy plays a pivotal role in regulating the antigen presentation and anticancer immune response by memory T cells and natural killer (NK) cells. 33 The deletion of ATG5 has been reported in NK cell malignancies, indicating that autophagy plays a crucial role in innate lymphoid cell (ILC) and NK cell development as well as tissue homeostasis. 34 Possible mechanisms include mitochondrial stress, excessive ROS production, and cell death. Similarly, ATG7 or ATG3 deletions have also been reported to impede T/NK cell development and deregulate the hematopoietic system, resulting in defective cells with high apoptotic activity. 34
Tumor progression
Autophagy is up- or down-regulated in many tumors. The role of autophagy in a particular tumor depends upon the tumor type and stage of disease. 35 Several studies have reported that autophagy is essential for cancer (stem) cell maintenance in various types of cancers.36–38 Kondo et al. 39 reported that autophagy is responsible for the survival of apoptosis‐defective tumor cells under chemo- or radio-therapy-induced metabolic stress. Indisputably, several reports suggest that hypoxia, commonly observed in solid tumors, leads to the activation of autophagy in cancer cells and controls glucose uptake via the regulation of glucose transporter 1 (GLUT1) activity.40,41 On the other hand, decreased expression of key autophagy proteins, LC3B-II and Beclin-1, was observed in glioblastoma multiforme. 42 Reduced autophagy was also reported in rat primary HCC cells, compared to that in normal hepatocytes. 43
A study conducted by Macintosh et al. underscores the importance of autophagy in metastatic tumors. In this study, the researchers found that the knockdown of Atg12 in glioma cells decreases the metastatic behavior of glioma cells. 44 RAS-mutated cancer cells have been found to have an elevated autophagy. RAS is known to play an essential role in cell proliferation, survival, and metabolism.45,46 Interestingly, a few anti-apoptotic or pro-survival BCL-2 family members (BCL-2 and BCL-XL) suppress autophagy through interactions with Beclin-1, thereby contributing to tumor progression. BCL-2 can block Beclin-1 by inhibiting its interaction with PI3KCIII either via dissociation of Beclin-1/PI3KCIII complex or via direct suppression of the Beclin-1 activity. 47 Alternatively, caspases can break down Beclin-1, thereby inhibiting autophagy. 47
Targeting autophagy in cancer therapy
Numerous studies have reported that autophagy plays a critical role as a tumor suppressor and promoter. The modulation of autophagy is a promising strategy for cancer therapy. In addition, it can also be used to improve the efficacy of existing conventional therapies as a combinatorial treatment strategy with other anti-cancer drugs. Here, we describe various autophagy inhibitors and inducers that are currently being used in preclinical studies and a clinical setting.
Autophagy inhibitors
According to ClinicalTrials.Gov, 30 clinical trials targeting autophagy in cancer are currently underway, the majority of them focused on inhibiting this process (Table 1). 48 Class III PI3K inhibitors such as Wortmannin, SAR405, 3-methyladenine (3-MA), Viridiol, and LY294002 block the formation of autophagosome by inhibiting PI3K, which plays a key role in ATG protein recruitment. 49 These inhibitors have been shown to reduce tumor proliferation in various cancer types when administered in combination with radio- or chemotherapy. 50 Lysosomotropic agents such as Lys0569, chloroquine (CQ), hydroxychloroquine (HCQ), quinacrine, and monensin block lysosomal acidification, thereby preventing degradation of the autophagosomal cargo. CQ and HCQ are being used in several clinical trials to sensitize cancer cells to radio- and chemotherapy, and promote survival. 51 While p53 mutation status in patients may influence clinical trial results, the combinatorial therapy of CQ/HCQ with other anti-cancer agents has been found to be more effective than monotherapy. 35 Although some patients have shown dose-limiting toxicities, very few side effects have been reported. Interestingly, CQ is also able to induce autophagy by inhibiting mTOR. 51 While CQ and HCQ have reached the clinical trial stage, other lysosomotropic agents are still being investigated at a preclinical level. Another class of agents including bafilomycin A1 (Baf A1) and concanamycin (Con), prevents autophagosome-lysosome fusion by inhibiting vacuolar ATPase. Several studies have reported the induction of apoptosis in tumor cells when Baf A1 is combined with 3-MA and other anti-cancer agents. 50 Spautin-1 leads to degradation of VPS34 and PI3K complexes by inhibiting ubiquitin-specific peptidases 10 and 13. It has been shown to increase apoptosis in chronic myeloid leukemia cells through blockade of PI3K/AKT signaling. 49 Nanoparticle-mediated delivery of anti-cancer agents has been reported to enhance autophagy in cells, and CQ was found to promote localization of such encapsulated agents on their targets by preventing their accumulation in organs through inhibition of macrophage uptake. 49 Autophgy levels can be modulated depending upon the design of nanoparticles and method of synthesis. 52 Inhibitors of ATG4B (major cysteine protease) and ULK1 have been shown to reduce/inhibit autophagic flux and enhance the anti-tumor effects of chemotherapeutic drugs in vitro and in vivo. 53 Alternatively, the expression of key autophagy components can be blocked genetically using specific DNA/RNA analogs as well as microRNAs (miRNAs), and this cost-effective strategy has shown promise in vitro and in vivo.49,54
Currently ongoing autophagy-targeted clinical trials.
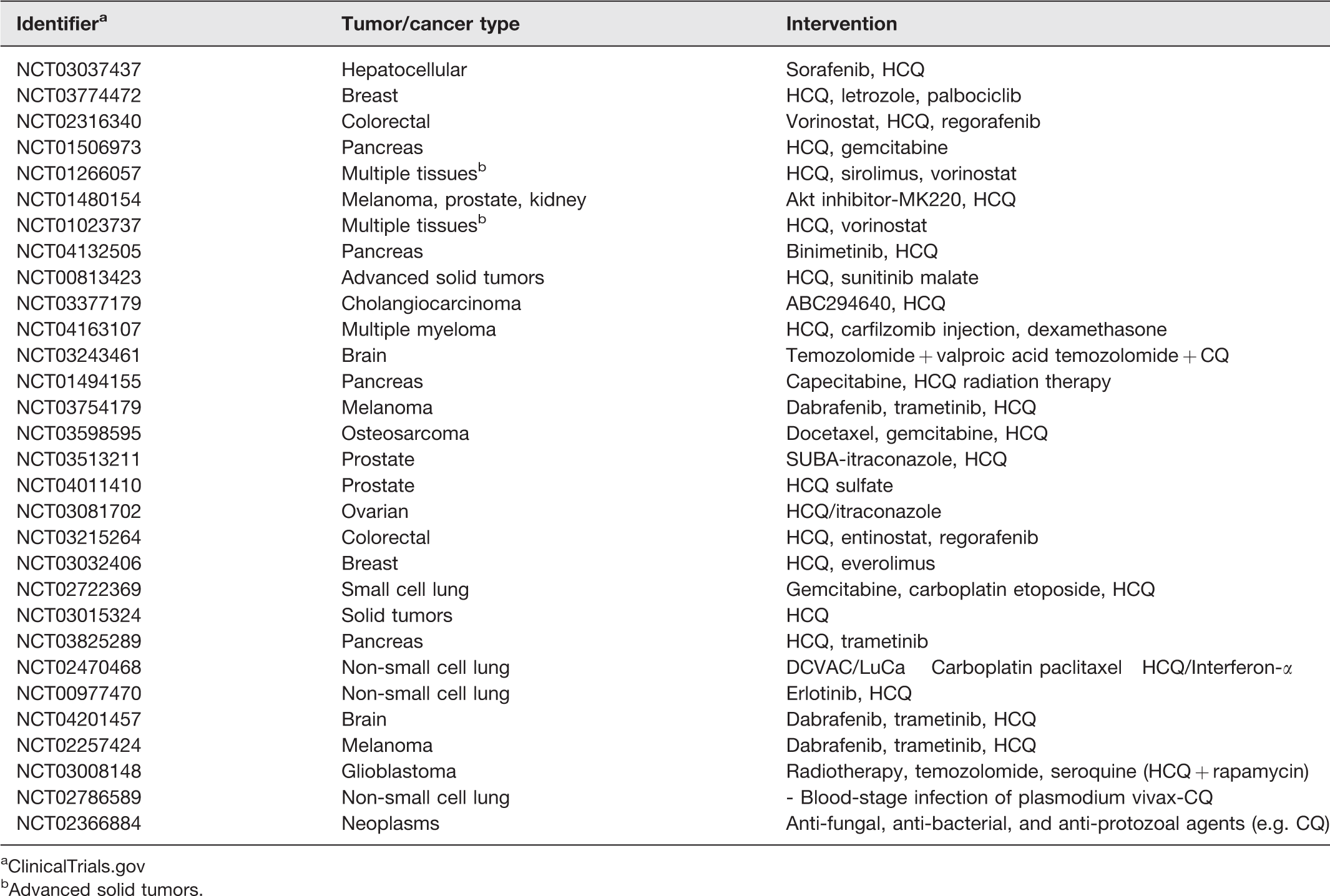
aClinicalTrials.gov
bAdvanced solid tumors.
As mentioned earlier, autophagy has multifaceted roles in cell immunity ranging from immune cell development, cross-presentation, and modulation of immune responses against cancer, which makes it an attractive target for use, as or in combination with, cancer immunotherapy, for improved clinical outcomes. Autophagy is currently being used as a therapy in the form of tumor vaccines such as p62-encoding DNA vaccine, Dribbles, and Bacillus Calmette-Guerin (BcG/cWs). 55 p62-encoding DNA vaccine (p62 is autophagy substrate) was found to inhibit tumor metastases in animal models. 55 Similarly, DRibbles, autophagosome-enriched vaccine, delayed melanoma growth and eliminated lung tumors in mice by stimulating cytotoxic immune responses. 55 BcG/cWs, another vaccine candidate, activated c-jun N-terminal kinase (JNK) and toll-like receptor (TLR) pathways, and induced autophagy in colon carcinoma cells when administered in combination with ionizing radiation. 55 Some studies also reported the efficacy of a dendritic cell-based vaccine in eliminating mouse 4T1 tumor metastases. 55 The use of autophagy inhibitors such as CQ and 3-MA has been found to augment the anti-tumor effects of immunotherapy with high dose interleukin-2 and interleukin-24, respectively. 56 Overall, these studies emphasize the role of autophagy as cancer immunotherapy.
Autophagy inducers
Autophagy acts as an alternative mechanism of death for cancer cells that escape apoptosis. mTOR inhibitors such as rapamycin and its analogs (e.g. temsirolimus, everolimus, ridaforolimus) induce autophagy through the formation and binding of FKBP12-rapamycin protein complex to mTORC1. 49 While rapamycin is able to inhibit cell proliferation by sensitizing cancer cells to radiotherapy, it has been used in very few clinical studies due to its poor stability and off-target effects. Cisplatin, a popular anticancer drug, was also found to inhibit mTOR signaling in in vitro endometrial cancer models. Likewise, paclitaxel induced autophagy in cancer cells, possibly though ROS generation and was accompanied by p62 downregulation. 54 Tyrosine kinase inhibitors such as sorafenib, imatinib, gefitinib, lapatinib, erlotinib, and vandetanib not only induce autophagy in cells but also impart resistance to death. Imatinib led to tumor cell death in vitro and in animal models. 49 Although sorafenib increased survival of progressed HCC patients, it failed to improve the overall survival rate, when administered in combination with mTOR inhibitor, everolimus, possibly due to drug resistance. 35 Histone deacetylase (HDAC) inhibitors such as vorinostat and valproic acid have been shown to induce autophagy. miR-101-3p, a type of miRNA, was found to enhance autophagy and reduce the expression of enhancer of zeste homolog 2 (EZH2), an enzyme involved in histone methylation, in endometrial cancer cells. 54 BH3 mimetic drugs induce autophagy by disrupting interactions between Beclin-1 and BCL-2. 51 Besides, several compounds such as arsenic trioxide, resveratrol, curcumin, ginsenosides, Fucus vesiculosus extracts, triterpenoids echinocystic acid, isoliquiritigenin, Itraconazole, and allicin have been reported to activate autophagy in cancer cells.49,54 More recently, cannabinoids have emerged as autophagy-inducing compounds and were shown to inhibit tumor growth in melanoma, HCC, and glioma cells. 57 A few studies have reported autophagy induction as a way to improve the efficacy of cancer immunotherapy since dying tumor cells could serve as potent immunogens. 16
Conclusion
Autophagy acts as a double-edged sword in cancer and some considerations should be taken into account before determining optimal intervention targeting the process. Although several clinical trials are ongoing, we are still unable to recognize the patient cohort that may benefit from treatments targeting autophagy. There is also an urgent need to develop more potent and specific autophagy inhibitors as well as biomarkers. Another caveat is that these inhibitors only work against tumors that are dependent on autophagy (RAS mutants) for their survival and it is difficult to distinguish such tumors from others that proliferate even in the absence of autophagy. Additionally, mutations like BRAF have been found to predispose the tumors to autophagy inhibition and targeting such tumors may overcome their resistance to chemotherapy. Currently, we are unable to find out whether autophagy modulation worked in vivo or whether it is actually targeting a particular disease and not adversely affecting other healthy organs. 16 The effect of such interventions seems to be cell-type specific. Therefore, it is as important to consider the differences in tumors originating from different tissues, as it is to study conserved underlying signaling pathways. The chosen intervention must be personalized to each patient for an effective treatment/cure.
Footnotes
AUTHORS’ CONTRIBUTIONS
MPJ, AV, and PG contributed to the conception, writing, and discussion of this review manuscript. The final version of the manuscript was approved by all authors.
Declaration OF CONFLICTING INTERESTS
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article..