
Abstract
Heart transplantation continues to be the gold standard clinical intervention to treat patients with end-stage heart failure. However, there are major complications associated with this surgical procedure that reduce the survival prognosis of heart transplant patients, including allograft rejection, malignancies, infections, and other complications that arise from the use of broad-spectrum immunosuppression drugs. Recent studies have demonstrated the use of mesenchymal stem cells (MSCs) against allotransplantation rejection in both in vitro and in vivo settings due to their immunomodulatory properties. Therefore, utilization of MSCs provides new and exciting strategies to improve heart transplantation and potentially reduce the use of broad-spectrum immunosuppression drugs while alleviating allograft rejection. In this review, we will discuss the current research on the mechanisms of cardiac allograft rejection, the physiological and immunological characteristics of MSCs, the effects of MSCs on the immune system, and immunomodulation of heart transplantation by MSCs.
Impact statement
This review is centered on the interactions between mesenchymal stem cells (MSCs) and immunological rejection after heart transplantation. The use of MSC as a new immunosuppressive agent has the potential to improve the survival of patients after heart transplantation. Understanding the process of rejection and the mechanism of protection provided by MSC is important for designing relevant studies. Furthermore, latest relevant results from rodent in vivo studies have been summarized in this review. Therefore, this review will not only provide useful information to guide basic research, but also discuss the potential clinical use of MSC.
Introduction
Since human heart transplantation was first performed by Dr Barnard in 1967, the procedure has proven to be the most effective and final treatment for end-stage heart failure. 1 However, the prognosis of heart transplant patients remains poor. Immune rejection remains to be the most significant challenge for heart transplant recipients despite the breakthrough use of immunosuppressive agents. While this treatment is effective against acute rejection, it is less so for chronic rejection. 2 Moreover, long-term use of nonspecific immunosuppression agents, such as Mycophenolate mofetil and Tacrolimus, are associated with many fatal complications. These include infections, malignancies, accelerated cardiovascular disease, metabolic complications, and direct toxic effects on transplanted tissues, all limiting the median survival of patients following heart transplantation procedure to under 12.5 years.3,4 Therefore, it is critical to find new methods to both reduce acute immune rejection and replace the long-term use of nonspecific immunosuppression agents.
In recent years, several studies have revealed unique properties of mesenchymal stem cells (MSCs) that regulate the immune response.5–7 Some of the novel regulation of MSCs to inhibit the immune rejection response occur at different levels, including the silencing of the cell activation of both the innate and adaptive immune systems and promoting a cardioprotective response from other cells. Due to their low immunogenicity, attributed to low expression of human leukocyte antigen major histocompatibility complex I (MHC I), and a lack of MHC II molecules and co-stimulatory antigens (such as CD80 and CD86), MSCs-based cell therapy promises several advantages over other cell-type based therapies against cardiac allograft rejection. Therefore, MSCs are considered immunologically safe for use in allogeneic clinical therapies. 8 Moreover, MSC-based cell therapy was shown to reduce necrosis, apoptosis, fibrosis, while promoting angiogenesis, therefore enhancing the overall cardioprotection and reducing the damage caused by immunological rejection with an increase in survival time of the transplanted heart. Thus, MSCs have significant potential to develop into the next novel preventative cell-based therapy for patients suffering from immunological rejection.
In this review, we provide a current synopsis of the alloimmunity response that accompanies heart transplantation and discuss the protective immunomodulatory function of MSCs on the innate and adaptive immune systems, and review the efficacy of MSC-based therapies in experimental models of heart transplantation.
Physiological and immunological characteristics of MSCs
Identification of MSCs
MSCs are a heterogeneous subset of non-hematopoietic cells that are characterized by their capacity to differentiate into tissues of mesodermal lineages. Although there are no unique cell-specific markers for MSCs, minimal criteria have been provided by The International Society for Cellular Therapy to define MSCs. Briefly, in vitro, cells with the ability of adherence to plastic under standard culture conditions, expressing cluster of differentiation (CD) genes such as CD105, CD73, and CD90, but not expressing CD45, CD34, CD14, CD11b, CD19, CD79, and HLA-DR genes. These cells have also been described to have the capacity to differentiate into chondrocytes, osteoblasts, and adipocytes in vitro. 9 MSCs can be obtained from several tissue sources, including bone marrow, umbilical cord blood, Wharton's jelly, amniotic fluid, adipose tissue, and dental pulp. 10
Immunological characteristics of MSCs
MSCs have the ability to inhibit the immune response by regulating several immune cell lineages at different levels; meanwhile, they show extremely low antigenicity allowing to escape alloreactivity. 11 Altogether, utilizing MSCs prove to be a potential cell-based therapy against cardiac allograft rejection. The most significant advantage of MSC therapy compared to the use of broad-spectrum immunosuppressive agents is their dynamic and specific regulation of the immune system. More specifically, broad-spectrum immunosuppressive agents protect allografts from rejection by nonspecific suppression of the total immune system; therefore, these agents not only stop the transplant rejection but also effectively shut down the immune surveillance of tumors and infections. Hence, transplant patients suffer from post-surgical health risks due to malignancies and severe infections when they are on broad-spectrum immunosuppressants for the rest of their lives.
In comparison, the immunomodulatory functions of MSC are dependent on and adaptive to the inflammatory microenvironment. 12 Preclinical studies have shown that tumor immune responses are not compromised by MSCs injection treatment. 13 Furthermore, MSCs have been shown to have antibacterial capabilities mediated by direct secretion of antibacterial factors or indirect activation of innate immune effector cells. 14 Together, these immunological properties of MSCs confer their bio-effectiveness and biosafety in comparison to broad-spectrum immunosuppressive agents.
MSCs alleviate innate immune response of cardiac allografts
MSCs protect cardiac allografts from ischemia-reperfusion injury
Currently, ischemia-reperfusion (I-R) injury remains as an inevitable part of organ transplantation. Following transplantation, the damage-associated molecular patterns (DAMPs) release, which is a potent activator of the innate immune response and correlates to the survival prognosis of the patient. 15 DAMPs include intracellular constituents such as nucleic acids or heat shock proteins that remain sequestered from the immune system during homeostasis but are released into the extracellular environment or exposed on the cell surface following cell damage.15,16 DAMPs also include extracellular constituents, such as matrix components that are modified in the context of cell stress or injury.15,17 During I-R injury, DAMPs are released and stimulate the innate immune system by activating pattern recognition receptors (PRRs), which are readily expressed and presented on the surface of most immune cells and some structural cells. 18 Toll-like receptors (TLRs), located at the surface and outer membrane of intracellular vesicles, are the principal member of PRRs. Activation of TLRs mediates a complex cascade of intracellular kinase activity, which later upregulates the transcription factor NF-κB and induce the expression of pro-inflammatory genes. 19 As a result of TLRs activation, the secretion of inflammatory cytokines, the recruitment of neutrophil, the maturation of antigen-presenting cells (APCs), and the upregulation of co-stimulatory molecules and the MHCs are all also elevated.18,20,21 Therefore, donor hearts begin to experience an allogeneic injury from the nonspecific immune response, with no resolution in sight due to continuous DAMP release upon further cell damage, and this vicious cycle of injury-response eventually leads to organ rejection.22–24
Hence, protecting the donor heart from I-R injury is a critical first step. On this front, several studies have shown the efficacy of MSCs against I-R injury.25–30 Korkmaz-Icöz et al. showed that in the presence of MSCs, in which the isolated hearts are perfused with a hypothermic histidine-tryptophan-ketoglutarate (HTK) solution supplemented by MSC conditioned medium, the donor hearts show greater preservation and gain resistance to I-R injury. 30 Compared to groups perfused with HTK only, the supply of MSCs-conditioned medium improved the cardiac function of the allografted heart. Moreover, the gene expression related to apoptosis, inflammation, and oxidative stress had all been downregulated. Similar results were also obtained on other solid organ preservation. For example, Daisuke et al. used MSCs therapy during ex vivo pig lung perfusion. They found that MSCs therapy can significantly protect the isolated pig lung from I-R injury with an evident decrease of apoptosis and inflammations. 31 Similar results have also been shown in liver transplantation trials. Even after 24 h of preservation at 4°C in University of Wisconsin (UW) solution, the portal transfusion of the MSCs ameliorated the injury of the liver graft after prolonged cold preservation and transplantation. 32 In light of these studies, MSC treatment of donor organs presents a critical improvement to the standard protocols of organ preservation and resistance to I-R injury.
MSCs affect maturation and differentiation of dendritic cells
Dendritic cells (DCs) are not only the APCs with the most potent antigen-presenting properties but also serve as an essential modulator of the cross-talk between the innate immune system and the adaptive immune system. Donor DCs mature immediately and migrate to the secondary lymphoid organs of the recipient through the lymphatic system under the activation of DAMPs-induced TLRs signaling.33,34 In the lymphatic organ, DCs present intact donor MHC–antigene complexes to host T cells, then provide efficient co-stimulatory signals like B7 molecules (namely CD80 [B7-1] or CD86 [B7-2]), CD40 to host T cells35,36 which is termed as the “direct presentation.” Subsequently, when the DCs from the donor’s heart are exhausted, DCs from the recipient take over. The antigen-presenting process mediated by recipient DCs is termed “indirect presentation.” Through direct presentation and indirect presentation, T cells are activated, which in turn caused the adaptive immune response (Figure 1).
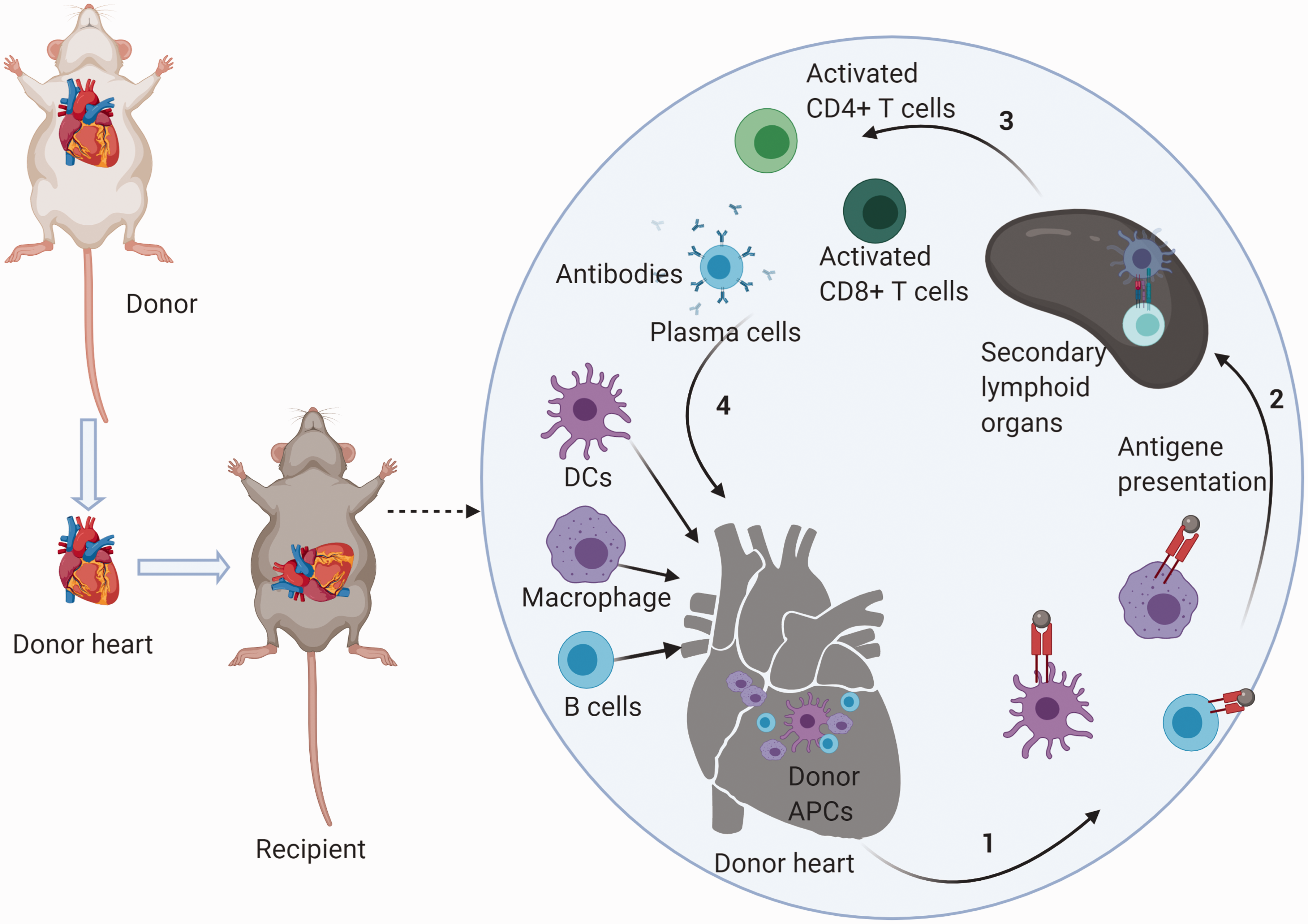
Schematic mechanism of cardiac allograft rejection. After heart transplantation, donor APCs present antigens to the secondary lymphoid organs of the recipient. In the lymphatic organs, APCs present intact donor MHC–antigene complexes to host T cells. T cells are activated by a combination of MHC–antigene complexes and efficient co-stimulatory signals. T cells then exert effects of adaptive immunity to cardiac allografts. When donor APCs are exhausted, recipient APCs take control. When recipient B cells present antigen to T cells, B cells are activated to plasmablasts. Plasmablasts then play a humoral immune response on the cardiac allografts. The effects of other innate immune cells are not shown in the figure.
DCs are only APCs that can activate inactivated naive T cells. The maturation of DCs is a necessary condition for their function to activate T cells. In this regard, MSCs have been shown to prevent the maturation and the migration of DCs. 37 Co-culture experiments with MSCs and DCs showed that the expression of co-stimulatory genes and antigen presentation of DCs are attenuated. 38 Interleukin-6 (IL-6) and macrophage colony-stimulating factor (M-CSF) secreted by MSCs have been reported to be involved in this maturation inhibition.37,39 Further studies have suggested that other soluble factors (such as prostaglandin E2 (PGE2)) may play a main role in the inhibition of DC maturation in addition to IL-6 and M-CSF. 40
Interestingly, DC inhibition through the paracrine factors secreted by MSCs was much weaker than the predominant regulation through MSC contact-dependent Notch signaling pathway. 40 MSC inhibitory actions regarding DC maturation have also been confirmed in a heart transplantation in vivo study. 41 Altogether, MSCs negatively influence DCs (the most significant APC), and prevent DCs from presenting antigens while inhibiting their ability to induce T-cell response. 42 Additionally, Zhao et al. have suggested that MSCs can induce differentiation of mature DCs into a distinct regulatory DCs (DCreg) population. 43 These regulatory DCs are known to promote anti-inflammatory effects on T cell activation, which result in CD4+CD25+Foxp3+ Treg cell generation from CD4+CD25-Foxp3− T cells. In a rat heart transplantation study, CD45RB+ DCs, which were induced by bone marrow-derived MSCs, promoted regulatory T cell (Treg) production and alleviate rejection injury in vivo. As a result, the survival time of cardiac allografts has been prolonged. 44 In summary, the anti-inflammatory effects of MSCs on DCs play a significant role in MSC-mediated immune tolerance induction.
MSCs polarize macrophages to M2 macrophages
Although the classification of macrophages is still controversial, most studies suggest that macrophages have two main subtype lineages, namely M1 and M2 macrophages. M1 macrophages are regarded as pro-inflammatory subtype of polarized macrophages. Hence, M1 macrophages secrete cytokines, such as IL1β, IL-6, IL12, tumor necrosis factor-α (TNF-α), which promote inflammation. M1 macrophages can be obtained by polarizing M0 macrophages (naïve) using lipopolysaccharides, or INF-γ (which are secreted by Th1 cells). 45 M2 macrophages are produced from M0 induced by IL4, IL13 (which are secreted by Th2 cells), as well as IL10 (which is secreted by Treg) and are considered the anti-inflammatory subtype of macrophages. Cytokines released from M2 macrophages, including transforming growth factor β (TGF-β) and IL-10, contribute to the anti-inflammatory response. These cytokines alleviate allograft immune injuries and facilitate tissue repairs. 46 Although macrophages are part of the APC family, like DCs, presenting donor antigen to the lymphocytes of the recipient, M2 macrophages do not express MHC-II molecules on their surface. 47 Thus, the skewing of M1 to M2 polarization is considered another feasible approach to regulate alloimmune rejection.
The effects of MSCs on macrophages reported in the literature are mostly reflected in terms of the enumeration on the polarization of macrophages. With the modulation of MSCs, macrophages are skewed toward M2 macrophage production. Many cytokines secreted by MSCs, such as IL10, PGE2, stanniocalcin-2, have been identified in this process.43,48–50 It is worth noting that the influence of MSCs on other immune cells also feeds back into the macrophage polarization. For instance, the effect of MSCs on the differentiation of Th2 cells and Treg indirectly contributes to M2 polarization. Gao et al. demonstrated the ability of MSC on macrophage polarization in vivo on a mouse heart transplantation model. 51 They suggested gene-modified MSC, such as soluble fibronectin-like protein 2 (sFgl2) overexpressing MSCs, therapy had a better benefit than wild type MSCs. This is of great importance, highlighting the potential of enhancing the MSC derived therapeutic response through further genetic modifications.
MSCs inhibit cytokine production and proliferation of natural killer cells
Natural killer (NK) cells are another central component of the innate immune system that recognize and attack allografts with a combination of MHC class I recognition and pro-inflammatory factor secretion. Once NK cells are activated, cytotoxic molecules are secreted without the need for priming by APCs. In addition, cytokines secreted from NK cells (such as TNF-α and IFN-γ) are involved in DCs maturation and Th1 cell polarization.52,53
Similar to previous findings, the proliferation of NK cells is significantly inhibited by co-culture with MSCs. 54 It is shown that MSCs are a potent inhibitor to activating receptors (e.g. NKp30, NKp44, and NKG2D) of NK cells, and this inhibition effect is further associated with impaired cytotoxic activity and cytokine production. Specifically, PGE2 and indoleamine-pyrrole 2, 3-dioxygenase (IDO) secreted by MSCs are suggested to mediate this process. 55 Recent studies reported that MSCs enhance the ability of IL-2/IL-12/IL-18-stimulated NK cells to secrete IFN-γ, which is a pro-inflammatory factor.56,57 However, it is important to mention that MSCs most likely play an anti-inflammatory role through NK cells on a limited NK cell-mediated inflammatory cases.
MSCs alleviate the adaptive immune response of cardiac allografts
The effects of MSCs on T cell activities: MSCs suppress T cell proliferation
Upon transplantation, naive T cells in host lymphoid organs quickly activate and proliferate due to the presence of MHC-antigene complexes and efficient co-stimulatory signals provided by APCs. It is noted that CD4+T cells, usually equipped with a helper function, are only able to recognize the donor peptides in the context of MHC class II molecules, which are presented on the surface of APCs. In contrast, CD8+T cells more often play cytotoxic roles and are activated by MHC I molecules expressed on nucleated cells (including donor APCs).
Several studies have shown that MSCs are potent suppressors of T-cell proliferation. In one example, Di Nicola et al. found that MSCs can inhibit both CD4+T cells and CD8+T cells proliferation in the mixed lymphocyte reaction system, and this inhibition is dose-dependent (positively associated with the total number of MSCs). 58 Furthermore, Sarah et al. demonstrated that the inhibition of T cell proliferation depends on the arrest of T cells in the G0/G1 phase of the cell cycle. 59 Interestingly, when MSCs were removed from the mixed culture system and restimulated with the cognate peptide, T-cells failed to proliferate but produced IFN-gamma. To date, many cytokines have been implicated in the proliferation inhibiting process of MSCs on T cells, including TGF-β1, hepatocyte growth factor (HGF), 58 IDO, 60 PGE2, 61 and NO. 62 However, Di Nicola et al. demonstrated that the paracrine effect of MSCs does not describe the entire mechanism by which they inhibit T-cell proliferation. 58 Comparing “non-contact” system, “contact” co-culture systems using MSCs with T-cells showed a stronger proliferation inhibiting.
MSCs affect the differentiation of CD4+ T cells
Activated CD4+ T cells can be classified into four main categories, including Th1, Th2, Th17, and CD4+Tregs. Th1 cells secrete pro-inflammatory cytokines (such as INF-γ/TNF-α) that can stimulate inflammatory responses in the transplanted heart by immune cell recruitment, and further promote activation of alloantigen-specific T-cells. 63 Th2 cells can trigger an antibody-mediated rejection and promote the involvement of eosinophils through the secretion of IL-4, IL-5, and IL-13. 64 Although acute rejection is typically viewed as a T cell-mediated process, and alloantibodies are considered a valuable treatment of chronic rejection, several studies have indicated that antibodies also serve a vital role in some cases of acute rejection.65–67
While IL17 is a well-known pro-inflammatory cytokine that is produced by Th17 cells, recently, it is shown that gamma delta T cells rather than CD4+ and CD8+ T cells were the key IL-17 producers in the allografts. 68 IL-17 recruit the immune cells, such as monocytes and neutrophils, to the site of inflammation and is crucial for the acceleration of acute rejection.69–71 Moreover, IL-17 has been identified to suppress Treg expansion.68,71 CD4+ Tregs are regarded as a significant part of donor heart tolerance. 72 CD4+ Tregs express the transcription factor FoxP3 and are known to suppress the alloreactivity through modulation of antigen presentation, production of anti-inflammatory cytokines, and competition and cytolysis of effector T cells.
In addition to affecting T cell proliferation, MSCs also affect T cell differentiation. Many studies have shown that MSCs are able to suppress the formation of Th1 and Th17, which are essential for the activation of cytotoxic T-cells and the boost of phagocytic capacity of neutrophils and macrophages.73–75 MSCs also contribute to the formation of Th2 cells. 76 As mentioned above, Th2 cells contribute to B-cell mediated rejection. However, in the presence of MSCs, B-cells proliferation in response to CD40L and IL-4 expressed by Th2 is also inhibited. 59 Thus, in this specific example, the ratio of Th2 to Th1 increased by MSCs is seen as a protective effect in allograft rejection setting. More importantly, MSCs are shown to prompt the production of Tregs via the secretion of IDO, 77 TGF-β1, and PGE2 78 by MSCs. Equally, the regulatory effect of MSCs on APCs is treated as a therapeutic target to indirectly control the mechanism to induce Treg production.79,80
Most in vivo studies using heart transplantation models attribute the cardioprotection of MSCs to the Tregs production and Th1/Th2 skewing.41,44,81–85 However, whether Tregs production via MSCs in vivo is yet to be explored by most authors. 86 Several outstanding questions including the influence of administration time, administration dose, and administration route of MSC on CD4+ T cell differentiation and fate in a heart transplantation model should be considered.
MSCs affect the formation and lysis function of CTLs, induce CD8+ regulatory T cells
CD8+ T cells activate into cytotoxic T lymphocytes (CTLs) with the assistance of CD4+ helper cells and banding of MHC-1 molecules presented by donor cells such as donor APCs. These cells then migrate into the donor’s heart, and lyse allogeneic recognized target cells. Specifically, CTLs release membrane-damaging proteins from intracellular granules (such as perforin and granzyme 87 ) and recruit other effector cells to the donor’s heart. Perforin polymerizes in the lipid bilayer of the donor tissue cell plasma membrane, creating a sizeable aqueous pore that allows other T-cell enzymes to enter into the target cell. Therefore, granzyme, as a serine protease enzyme in T-cell granules, enters the target cells through the pore, and then proteolytically cleaves and activates caspases that induce target cell apoptosis. In addition, activated CD8+ T cells also express FasL on their surface. When the FasL binds to their Fas receptor, which is expressed on target cells, Fas/FasL pathway will be activated. Fas/FasL signaling pathway is another critical mechanism that is directly utilized by CD8+ T cells to induce apoptosis. 88
MSCs are suggested to have a strong ability to inhibit CTLs formation in vitro. 89 In the presence of MSCs, the lysis function of CTLs on target cells has been significantly inhibited. The effect of MSCs on CTLs is mainly mediated by their paracrine function. PGE2, IDO, and TGF-β are suggested being involved in this process. 90 Furthermore, there is evidence that shows the correlation between MSCs and CD8+ regulatory T cells increase.91,92 Similar to CD4+ regulatory T cells, CD8+ regulatory T cells can inhibit the immune response of CD4+ T cells by several pathways. 93 CD8+ regulatory T cells have several subtypes with different surface markers. CD8+CD28– regulatory T cells is a widely recognized subtype of CD8+ regulatory T cells. Liu et al. reported that MSCs increase the proportion of CD8+CD28− T cells by reducing their rate of apoptosis. 92 MSCs also enhance CD8+CD28− regulatory T cells’ ability to hamper naive CD4+ T-cell proliferation and activation.
Although the effects of MSCs on CD8+ T cells may provide novel therapeutic strategies for heart transplant tolerance, there is a big gap of knowledge for the relevance of this mechanism in an in vivo laboratory model.
The effects of MSCs on B cells activities
B cells are one of the three primary subtypes of APCs and differ from DCs and macrophages with their capability of the secretion of specific antibodies. B cells can specifically recognize antigens from the donor’s heart with B cell receptors (BCR), which is formed by membrane-bound IgD and an IgM receptor in addition to other membrane molecules. Although such recognition and binding is the first step to initiate the process of activation, it is not enough to fully activate the function of B-cells. The assistance of other factors (including complement and CD4+ T helper cells) is required for B-cell activation. To complete the activation, B-cells endocytose and process the donor antigen bound with BCR, then present them to CD4+ T cells in the form of MHC II-antigene complexes. In return, activated T cells will bind the CD40 molecules on B cells surface with their ligand CD40L. CD40–CD40L interaction is a strong co-stimulatory signal for B-cell proliferation and differentiation.
Moreover, cytokines produced by activated CD4+ T helper cells will also add to a more fully developed B-cell response. During B cells activating, soluble Igs that is secreted from the cells are elevated, and membrane-bound Igs that are attached to the surface of a B cell are gradually decreased. Then, most activated B-cells differentiate into antibody-secreting cells (also named Plasma cells), which mediate specific antibody targeted to an antigen expressed on the cells from the donor heart. The form of antigen–antibody complexes activates complement cascades, ultimately transforming the endothelial cell surface from an anticoagulant condition to a procoagulant status. Therefore, microthrombus can be formed from the tiny blood vessel, and finally cause the closure.
Furthermore, activation of complement cascades also initiates the development of the membrane attack complex, which contains a series of pores in the cell membrane that bring about cell death in the donor’s heart. Coincidentally, some of the products from the complement system (like C3a and C5a) are puissant triggers of mast cell degranulation and inflammations. Even without assistance from the complement system, Fc receptor-bearing effectors can recognize and kill antibody-coat target cells expressing an allogeneic antigen on their surface. These Fc receptor-bearing effectors include follicular DCs, NK cells, macrophages, neutrophils, eosinophils, basophils, and mast cells that simultaneously exert their immune functions on the donor’s heart.
MSCs have been reported to influence B-cell proliferation, plasmablast formation, and promote the induction of regulatory B-cells (Bregs).59,94 Like Treg, Breg is an immunosuppressive subtype of B cells. The secretion of IL10 by the Breg was shown to convert effector CD4+ T cells into Foxp3+ Tregs. 95 Interestingly, it is reported that the stimulatory effect of MSCs on Breg formation and IL-10 production is mediated by direct cell–cell contact mechanism and not through a paracrine signaling.96,97 Although B-cells may directly be influenced by MSCs to alter their characteristics of proliferation, antibody production, and other immune cells (e.g. T-cells and DCs), it is equally possible that MSCs play an indirect regulatory role.98,99 Unfortunately, it is still unknown whether MSC has any effect in chronic rejection of heart transplantation by exerting related effects on B cells in vivo experiments.
Challenges for use of MSCs in cell therapy
There are several challenges to characterize the immunomodulation involved in MSCs-based cell therapy. Firstly, the heterogeneity of MSCs related to donor origin, cell development stage, and expansion protocol are major issues that restrict the therapeutic benefits of MSCs. 100 For instance, culture conditions and expression of miRNAs can modify the immunomodulatory function of MSCs. Secondly, cell distribution to the targeted tissues would be limited after delivery. The majority of MSCs could be transferred to organs with a high vessel density (especially the lung) through the circulation system. 101 With the increase in infused cells, a risk of occlusion in microvessels is raised. Thirdly, the long-term survival and effectiveness of infused cells are still a concern. Although MSCs have an extremely low antigenicity, repeated administration of MSCs may cause the production of antibodies. 102 Finally, although anti-tumor immune responses are not compromised by MSCs, the potentials of ectopic tissue formation and malignant transformation are still concerned. Importantly, these challenges would be addressed with the use of MSCs.
Compared to cell infusion, delivery of exosomes or extracellular vesicles (EVs) derived from MSCs might be an attractive strategy for development of a cell-free therapeutic. As a type of nano-level membrane particle, the secreted exosomes/EVs can pass through most physiological barriers and have been considered a major contribution to the therapeutic effect of MSCs via a paracrine pathway. It remains unknown whether the exosomes have similar capacities as MSCs for alleviating cardiac allograft rejection in specific conditions. However, as mentioned above, the immunomodulatory effects of MSCs not only rely on several growth factors or cytokines, but cell–cell contact is also involved in the procedure. Furthermore, MSCs’ immunomodulatory effects are not static in response to the varying microenvironment. Therefore, the components of exosomes/EVs would be complex and their dynamic changes may lead to various consequences in different settings. To optimize this cell-free approach, the isolation or manufacture of exosomes/EVs should be developed in a GMP condition and their biological features must be clearly defined prior to clinical use. Additional investigation is needed to determine whether exosomes or EVs can completely replace MSCs in the treatment of cardiac allograft rejection in the future.
Conclusions
In this review, we provide a summary of immunomodulatory effects of MSCs for the treatment of cardiac allograft rejection. Although the immunomodulatory effects of MSCs observed from in vitro experiments reveal exciting therapeutic possibilities for MSC-based cell therapy (Figure 2), conflicting data in heart transplantation models with limited reproducibility render the application of this therapy experimental at best. However, there is a general agreement on some benefits from MSC cell therapy along with organ transplantation against allograft rejection and tolerance induction. A few studies have indicated that MSCs are more harmful than helpful and actually promote rejection.103,104 The immediate goal at the moment seems to be the elimination of acute rejection and the reduction of the use of immunosuppressor agents, which may help induce immune tolerance in clinical cases.
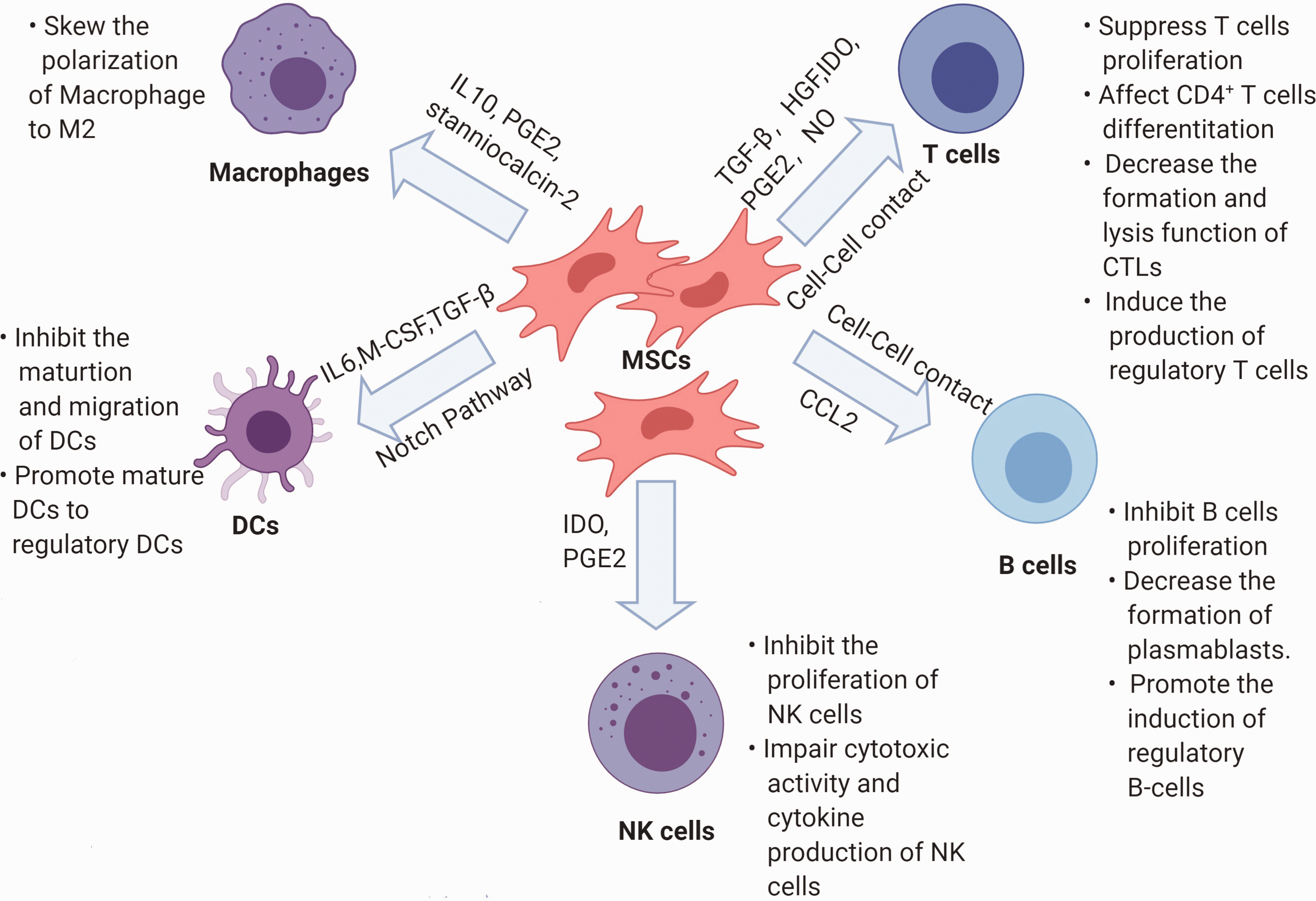
Schematic representation of modulation in immune cells by MSCs.
Currently, studies need to focus on verifying the ability of MCSs to reduce immune response and explore the underlying mechanisms in heart transplantation models. As mentioned above, the immunomodulatory effects of MSCs are never static and depend on the microenvironment. Therefore, experiments need to identify, detect, and control the changes in the microenvironment while developing a reproducible heart transplant surgery method. Since MSCs express several TLRs,105–107 the types of DAMPs released either due to ischemia/reperfusion injury, inflammations, hypoxia all may modify the patterns and functions of TLRs expressed on MSCs, and therefore impact the interactions between MSCs and immune systems.106,108–110
Similarly, the source and state of MSCs used in studies need to be better defined and controlled in order to parse out the difference in their function and response in vivo. Therefore, there are so many outstanding questions and concerns that need to be addressed regarding immunogenicity, mechanisms of immunomodulation and differentiation of MSCs, administration time, administration route, the dosage of administration, indications and contraindications of administration, and the long-term safe use of these cells must be assessed before MSCs can be utilized as a suppressive treatment in a clinical setting.
Footnotes
AUTHORS’ CONTRIBUTIONS
All authors (ZW, JL, WH, LJ, CP, XG, PA, OK, MX, YG) participated in the design and interpretation of the manuscript. JL, WH, LJ, and PA contributed to manuscript preparation. ZW drafted and wrote the manuscript. YW drafted and critically revised the manuscript. CP, OK, and MX carefully edited the manuscript.
DECLARATION OF CONFLICTING INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship, and publication of this article.
FUNDING
The authors disclosed receipt of the following financial support for the research, authorship, and publication of this article: This work was supported by the National Institutes of Health (RO1 HL143490, RO1 HL136025 (YW)). This work was also supported by the National Institutes of Health [RO1 HL148598-01A1, (OK)] and American Heart Association, Career Development Award, 18CDA34110117 (OK). Finally, AHA award:18CDA34110223(JL), AHA postdoctoral grant: AHA_20POST35200267 (PA) were also used to support this work.