
Abstract
Therapeutic interventions aimed at inducing fetal hemoglobin and reducing the concentration of sickle hemoglobin is an effective approach to ameliorating acute and chronic complications of sickle cell disease, exemplified by the long-term use of hydroxyurea. However, there remains an unmet need for the development of additional safe and effective drugs for single agent or combination therapy for individuals with β-hemoglobinopathies. Regulation of the γ-globin to β-globin switch is achieved by chromatin remodeling at the HBB locus on chromosome 11 and interactions of major DNA binding proteins, such as KLF1 and BCL11A in the proximal promoters of the globin genes. Experimental evidence also supports a role of epigenetic modifications including DNA methylation, histone acetylation/methylation, and microRNA expression in γ-globin gene silencing during development. In this review, we will critically evaluate the role of epigenetic mechanisms in γ-globin gene regulation and discuss data generated in tissue culture, pre-clinical animal models, and clinical trials to support drug development to date. The question remains whether modulation of epigenetic pathways will produce sufficient efficacy and specificity for fetal hemoglobin induction and to what extent targeting these pathways form the basis of prospects for clinical therapy.
Impact statement
Therapeutic options to induce fetal hemoglobin that are safe and effective are a high priority for treating individuals with sickle cell disease. Tissue culture, preclinical, and clinical studies provide evidence for targeting epigenetic mechanisms of reversing the γ-globin to β-globin gene switch during development. The goal of this review is to critically assess the role of epigenetic mechanisms in γ-globin gene regulation and discuss the question whether modulation of epigenetic pathways will produce sufficient efficacy and specificity for fetal hemoglobin induction to warrant future development for clinical therapy.
Introduction
Sickle cell disease (SCD) is one of the most common inherited blood disorders that affect more than 100,000 individuals in the United States and millions of people worldwide. 1 Fetal hemoglobin (HbF) is the major genetic modulator of the hematologic and clinical manifestations of SCD. For example, people with hereditary persistence of fetal hemoglobin maintain high HbF levels of 20–30% and have less symptoms and better clinical outcomes. 2 Thus, therapeutic interventions aimed at inducing HbF expression while reducing the concentration of sickle hemoglobin (HbS), are effective approaches to ameliorating complications of SCD. To achieve this goal, additional knowledge about the molecular and cell signaling mechanisms involved in regulating γ-globin transcription are required. Induction of HbF requires chromatin remodeling of histones at the γ-globin (HBG) and β-globin gene promoters.3,4 Epigenetic modifications including acetylation and methylation influence access of trans-acting DNA-binding proteins such as GATA1, TAL1, E2A, LMO2, LRF/ZBTB7, and LDB1, which facilitate DNA looping of the locus control region (LCR) with each globin promoter, to enhance gene transcription.5,6 Pre-clinical and clinical evidence described to date suggests that epigenetic modifications mediated by DNA methyltransferases (DNMTs), histone methyltransferases, histone demethylases, histone deacetylases (HDACs), and histone acetyltransferases (HATs) along with changing microRNA (miRNA) expression levels, may be involved in the γ-globin to β-globin switch. However, it remains unclear how important these epigenetic mechanisms are in HbF induction and whether drugs that target changes in epigenetic marks will provide sufficient efficacy and potency to achieve clinical utility. In this review, we will critically assess epigenetic mechanisms involved in globin gene regulation and discuss the question as to whether modulation of epigenetic pathways will produce sufficient efficacy and specificity for HbF induction to warrant expanded development for clinical therapy.
Food and drug administration-approved therapies for sickle cell disease
Disease-targeted treatment options for individuals with SCD were limited to hydroxyurea (HU) for over two decades. More recently, three additional agents were FDA-approved including Oxbryta® to inhibit HbS polymerization, Crizulizamab® to block interactions with P-selectin glycoprotein ligand 1 (PSGL-1), and Endari® to increase L-glutamine levels and reduce oxidative stress within sickle cells.7–9 After many decades of research, no other HbF-inducing agents have been FDA approved, creating an unmet need for additional drug development for SCD.
Epigenetic mechanisms of globin gene regulation
A broad spectrum of pharmaceutical agents has demonstrated the ability to induce HbF in tissue culture; however, their efficacy in preclinical animal models and clinical studies has not been demonstrated. Many of these molecules act independently of DNA sequence-based genetic mutations of the γ-globin gene. Experimental data suggest these mechanisms, termed epigenetics, including DNA methylation, histone acetylation/methylation and changing microRNA (miRNA) expression levels,10–17 alter transcription of the γ-globin gene resulting in reversal of the γ- to β-globin switch.10,18–21
DNA methylation
DNA methylation was the first described epigenetic signal postulated to play a role in globin gene regulation during development.22–28 In 1980, van der Ploeg et al. 23 analyzed the methylation status of the γ-globin promoter by digestion of DNA isolated from erythroid and nonerythroid tissues, with methylation sensitive restriction enzymes and Southern blot analysis. They were among the first group to establish a significant correlation between human γ-globin promoter methylation levels and gene transcription during development. 23 Shortly after, Busslinger et al. 28 showed that methylation of nucleotides located at positions −760 to +100 in the 5ʹ region of the γ-globin gene prevented gene transcription. 28 Early studies by Comi et al. in 198629 showed that treatment of adult erythroid cells with bromo-deoxyuridine, a nucleotide analog that inhibits DNA methylation, reactivated HbF expression. 29
Since the discovery of the role of DNA methylation in globin gene regulation, a large body of literature supports changes in methylation levels of the human HBB locus on chromosome 11, in contributing to hemoglobin switching mechanisms. 14 ,18,19,30–34 During developmentally regulated erythroid cell differentiation, the five globin genes (5-ʹε, Gγ, Aγ, δ, β-3ʹ) in the HBB locus become sequentially demethylated and transcriptionally active. 35 Specifically, in late gestation, the CpG dinucleotides of the γ-globin promoters are hypomethylated supporting gene expression until after birth with completion of hemoglobin switching by one year of age in normal infants.32, 36 This finding was further supported by Singh et al. 37 showing that γ-globin promoter hypermethylation in early fetal liver gradually demethylates as erythroid differentiation progressed over time. 37 Conversely, during adult erythropoiesis, the γ-globin gene promoter becomes hypermethylated and silenced by DNA methylation machinery,18,30–34 producing a 20-fold repression in gene transcription. 36
To achieve epigenetic regulation, DNMTs interact with various transcription factors that bind the γ-globin proximal promoter facilitating highly methylated CpG sites located at positions −256, −162, −71, −53, −50, +6, +17, and +5010,38 Additionally, chromatin regulators, including DNMT3A, the lysine methyltransferase SUV4-20 h1, the serine/threonine kinase CK2alpha, and components of the Nucleosome Remodeling and Deacetylase (NuRD) co-repressor complex, occupy the γ-globin promoter to repress its expression.39,40 It has been shown that silencing of DNMT1 in primary cultures of erythroid progenitor cells derived from CD34+ stem cells isolated from baboon bone marrow cells, increased levels of γ-globin mRNA and decreased DNA methylation of the proximal promoter. 41 These findings highlight the involvement of DNA methylation machinery and cofactors in regulating chromatin structure in the HBB locus and γ-globin gene silencing in adult-stage erythroid cells.
Transcriptional repressors of γ-globin gene expression
Genetic mapping and later genome-wide association studies (GWAS) discovered quantitative trait loci in the B-cell lymphoma/leukemia 11 A (BCL11A) gene, an XmnI variant upstream of the Gγ-globin gene, and the HBS1L-MYB intergenic region that were associated with inherited variations in human HbF levels.42–46 The C > T genetic variant in XmnI (rs7482144) located at −158 upstream of the Gγ-globin gene was correlated with disease severity in adults with β-thalassemia. 43 The BCL11A gene encodes a C2H2 type zinc-finger protein, which is a major transcriptional repressor of γ-globin during adult erythropoiesis.47,48 Inherited polymorphisms in BCL11A associated with HbF levels in hemoglobinopathy patients 49 provided the impetus for laboratory studies demonstrating BCL11A transcription factor as a major silencer of γ-globin gene expression (Figure 1). KLF1 was the first major transcription factor discovered to play a key role in adult β-globin gene activation during hemoglobin switching. 50 Later studies demonstrated the role of KLF1 in the γ- to β-globin gene switching process by directly activating BCL11A, 51 which indirectly repressed γ-globin expression.52–55 KLF1 also regulates many components of the cell cycle machinery during erythropoiesis.52,54,56,57
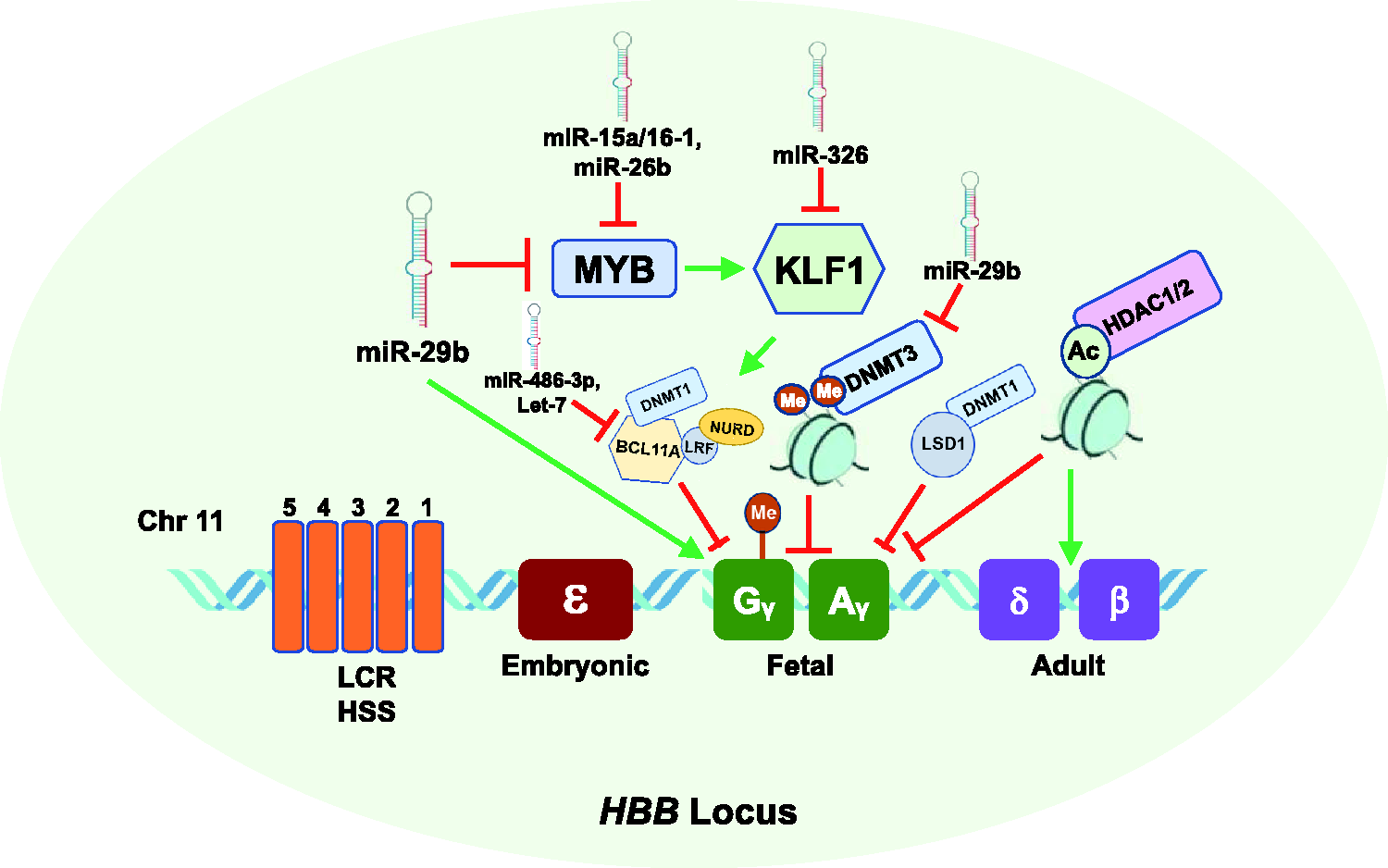
Effect of epigenetic mechanisms, miRNAs, and transcription factors on fetal globin gene expression during hemoglobin switching. Shown is the HBB gene locus on chromosome 11 which consists of the five developmentally regulated globin genes, including fetal γ-globin (green) and adult β-globin (purple). Silencing of γ-globin genes is mediated by transcriptional repressors MYB which activates KLF1, which in turn activates the repressor BCL11A which mediates transcriptional silencing of γ-globin. The γ-globin promoters become hypermethylated and silenced by DNMT1. The DNMT3 proteins, DNMT3A and DNMT3B, are required for long-term methylation of the γ-globin gene promoters and silencing during adult erythropoiesis. In addition, deacetylation by HDAC1/2 inhibits γ-globin expression and activates adult HBB expression. MiR-29b, a DNMT3 inhibitor, inhibits MYB expression resulting in γ-globin gene activation. Additional miRNAs that inhibit (red line) or induce (green line) HbF are shown. (A color version of this figure is available in the online journal.)
Additional studies support an indirect role of the oncogene MYB 58 in regulating γ-globin transcription through targeting BCL11A and the repressor TR2/TR4.38,54 MYB is highly expressed in immature hematopoietic cells and down regulated during erythropoiesis. 59 Overexpression of MYB transcription factor inhibits γ-globin expression in K562 cells, 58 and MYB silencing induces HbF expression in primary human erythroid progenitors. 60 In human genetic studies, the association of MYB with HbF levels was demonstrated using quantitative trait loci studies and subsequent functional assays.43,58,60 The leukemia/lymphoma-related factor (LRF), encoded by the ZBTB7A gene, is a ZBTB transcription factor that directly binds fetal γ-globin promoter and represses γ-globin gene expression during adult erythropoiesis through a NuRD repressor complex independent of BCL11A 61 (Figure 1).
Ten eleven-translocation dioxygenases
The TET enzymes are a family of TET methylcytosine dioxygenases that are involved in DNA demethylation. 62 We previously showed that the γ-globin gene transcriptional activator, NRF2, which modulated chromatin structure in the human HBB locus of β-YAC transgenic mice during development, interacted with TET3 to regulate γ-globin methylation. 16 Tet2 and Tet3 dioxygenases that catalyze formation of 5-hydroxymethylcytosine (5 hmC) and are expressed during early stages of erythroid differentiation were also shown to regulate γ-globin expression by mediating demethylation of the γ-globin promoter. 18 Conversely, Sp1 binding to the −71 to −34 sequence of γ-globin increased upon site-specific cytosine methylation, suggesting Sp1 is a potential repressor of γ-globin expression.63,64
Protein methyltransferases and demethylases
Another group of epigenetic modifiers include lysine demethylases (LSD), which are enzymes that can demethylate mono- and di-methylated lysines. 65 They are known to interact with BCL11A to influence γ-globin gene silencing in both murine and human adult erythroid cells. 66 Specifically, LSD1 associates with BCL11A through a complex containing the repressor element-1 silencing factor co-repressor-1 (CoREST) to mediate γ-globin gene silencing. 66 Supporting studies using RNA interference to silence LSD1 or the LSD1 inhibitor tranylcypromine support a role for LSD1 in γ-globin regulation.66,67 A study by Rivers et al. 68 demonstrated that oral administration of the LSD1 inhibitor ORY-3001 increased F-reticulocytes, γ-globin mRNA, and HbF synthesis in baboons and SCD mice. 68 However, there were concerns with potential adverse effects when tested in preclinical transgenic mice and primate models34, 67,68 (Table 1).
Summary of epigenetic therapeutics tested in clinical trials for sickle cell disease.
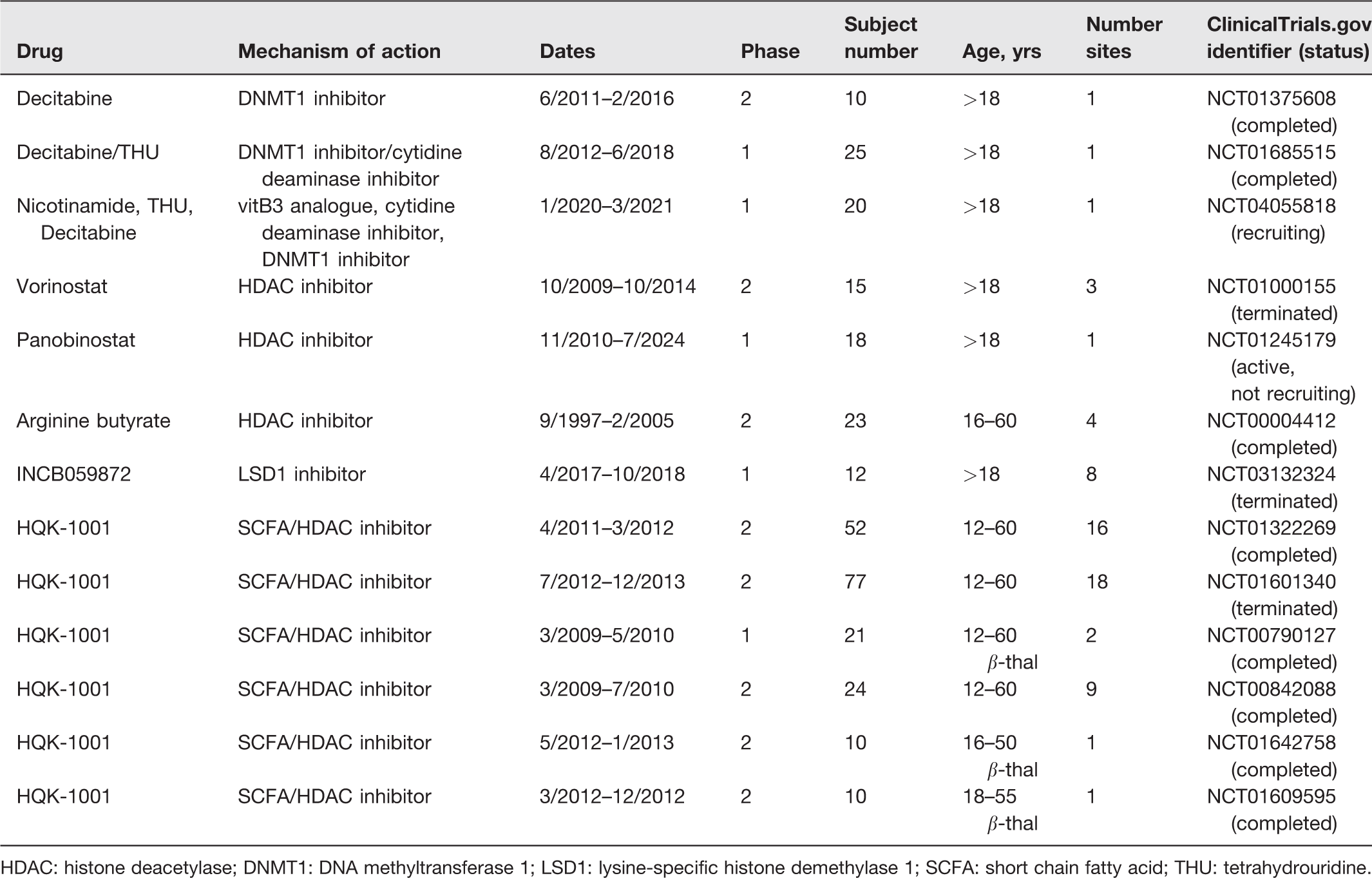
HDAC: histone deacetylase; DNMT1: DNA methyltransferase 1; LSD1: lysine-specific histone demethylase 1; SCFA: short chain fatty acid; THU: tetrahydrouridine.
Another group of proteins involved in globin gene regulation is the methylated DNA binding protein (MBD) family. The MBD family includes methyl CpG binding protein 2 (MeCP2), which interacts with DNA containing methylated CpG-rich sequences with high affinity. 69 MBD proteins regulate gene activity by modifying chromatin through recruitment of histone deacetylases and methylases to methylated DNA. 69 Our group and others have shown that MeCP2 and/or other MBD proteins are involved in γ-globin regulation in adult erythroid progenitors.15,63,70 Indeed, depletion of MBD2 produced a 10 to 20-fold increase in γ-globin expression in β-globin yeast artificial chromosome transgenic mice.71,72 Mechanistically, proteins in the MBD family recruit chromatin remodeling members of the NuRD co-repressor complex. The latter is an important epigenetic reader of DNA methylation that regulates gene transcription during normal development and neoplastic diseases15,70 through protein–protein interactions. Studies in primary adult erythroid cells showed that knockdown of the MBD2-NuRD complex increased the γ/γ + β mRNA ratio by 10-fold which correlated with an increase in HbF protein levels. 72 Other studies showed that the direct repeat erythroid-definitive (DRED) and BCL11A-NuRD co-repressor complexes, containing DNMT1 protein, were also involved in γ-globin gene silencing in adult erythroid cells.66,73
Collectively, these studies provide strong evidence of a critical role for DNA methylation and DNA methylation machinery in hemoglobin switching during adult erythropoiesis and highlight the potential of DNA methylation-targeted therapies for HbF induction. However, studies have shown that HbF levels of ∼30% with pancellular distribution are required to ameliorate SCD clinical severity. 74 Therefore, it is unclear as to what extent the increase in γ-globin transcription produced by repression of the MBD2-NuRD complex translates into clinical efficacy.
DNA methylation agents in HbF induction
Two DNMT inhibitor agents, namely 5-azacytidine and its analog decitabine, have been investigated for many years as HbF-inducing agents75–83 (Table 1). 5-azacytidine was first demonstrated to induce HbF in anemic baboons 84 and later in individuals with SCD and β-thalassemia.85–90 Decitabine and 5-azacytidine are both DNMT inhibitors that reactivate γ-globin expression via hypomethylation of the CpG site within the γ-globin gene promoter which allows binding of erythroid-specific transcription factors such as KLF1, BCL11A, GATA-1, and GATA-2 to chromatin sites in the HBB locus.18,51,91 Indeed, decitabine is a potent HbF inducer when given parenterally but oral administration leads to rapid degradation by intestinal cytidine deaminases.92,93 Furthermore, there are major concerns with adverse side effects, including neutropenia, thrombocytopenia, thrombophilia, or cytotoxicity from taking decitabine.94–96
More recent efforts have focused on the administration of tetrahydrouridine (THU) before decitabine to prevent rapid degradation in the liver 93 (Table 1). In fact, a recent clinical trial combining oral decitabine and THU showed promising results associated with increased HbF and total hemoglobin levels in persons with SCD. 31 In 2020, the US FDA approved the combination of oral cedazuridine, a synthetic nucleoside analog derived from THU, and decitabine (INQOVI) formulation for use in adults with myelodysplastic syndrome. 97 Supporting studies in acute myeloid leukemia patients and in non-human primate baboons showed that pharmacological inhibitors of DNMT, namely S110, 5-aza-2ʹ-deoxycytidine, and decitabine, induced high levels of HbF.18,84,98,99 A newer generation methyltransferase inhibitor, adenosine-2′, 3′ dialdehyde (Adox) was shown to induce γ-globin gene expression in human primary erythroid cells in culture. 100 The recent discovery of a novel orally bioavailable DNMT1-selective inhibitor, GSK3482364 which directly blocks DNMT1 enzymatic activity while sparing DNMT3A or DNMT3B, decreased γ-globin promoter methylation and increased HbF expression in adult human erythroid progenitor cells and SCD transgenic mice. 101 However, due to a limited number of therapies to treat SCD and concerns about adverse effects from decitabine and other DNA methylation inhibitors with known cytotoxic effects,94,95 there remains an unmet need for the development of novel non-cytotoxic agents to target DNA methylation machinery to induce HbF.
Histone modifications
Post translational histone modifications including histone acetylation and methylation are involved in the regulation of γ-globin gene transcription. 102 The developmentally regulated expression of the globin genes requires sequential interaction with the LCR, which consists of five DNA I hypersensitive sites (HSs), with the individual globin promoters. The process of histone acetylation is strongly correlated with an open chromatin conformation and increased gene transcriptional activity, whereas methylation of histone residues can either increase or decrease gene transcription, depending on the location and number of amino acid residues methylated. 102 For instance, formation of histone H3K4me3 is usually associated with transcriptional activation, while histone H3K9me3 and H3K27me3 are associated with inaccessible chromatin and gene silencing.103,104 The general process of histone acetylation is catalyzed by HATs which transfer an acetyl functional group via acetyl coenzyme A from one molecule to another. Increased histone acetylation is typically associated with chromatin opening resulting in the binding of transcription factors to activate gene expression, whereas histone deacetylation tends to condense chromatin into a tightly packed state, which prevents binding of regulatory factors.
Histone acetylation
Indeed, increased histone acetylation has been strongly associated with increased γ-globin gene expression. 13 Supporting studies have implicated the process of acetylation and methylation of lysine residues on histone tails in HbF induction in erythroid cells and preclinical animal models.10,105–108 Specifically, acetylation of H3 and H4 lysine residues and recruitment of RNA polymerase II to the γ-globin promoter region results in elevated HbF expression.12,109 Conversely, deacetylation of the lysine residues within the N-terminal tails of H3 and H4 histones has been observed to shift the balance of γ-globin gene expression resulting in gene silencing.110,111 The observation of ectopic expression of the protein deacetylase SIRT1 and SIRT activators (SRT2104 and SRT1720) increased γ-globin gene expression in primary human erythroid cells and K562 cells was surprising. 112 Interestingly, SIRT1 also increased binding of RNA polymerase II and acetylation of H4K16 at the γ-globin promoter, thereby enhancing gene transcription. 112 Another study reported that histone acetylation is dependent on binding of the erythroid-specific transcriptional activators GATA-1 and NF-E2 at the HBB locus which was not sufficient for γ-globin transcription. 113 However, Layon et al. 114 reported that GATA-1 alone was sufficient to direct chromatin structure reorganization of the HBB LCR and an erythroid pattern of gene expression in the absence of other hematopoietic transcription factors such as NF-E2. 114
The complexity of the relationship between histone deacetylation and globin gene regulation has been investigated in earlier studies by Perrine et al. 108 who demonstrated that the HDAC inhibitor, butyrate, significantly delayed the globin gene switch in sheep fetuses compared with controls. Similar studies found that specific histone acetylation patterns play a critical role in the developmental switch of the murine HBB genes. 106 Their findings, along with other supporting studies,106–108 led to the discovery of butyrate in silencing γ-globin activation in erythroid progenitors, preclinical models, and patients with SCD.106,107,115–117 In addition to butyrate, HDAC1 and HDAC2 have been further recognized as molecular targets mediating HbF induction through a chemical genetic strategy and RNA interference. 118 Moreover, our group showed that butyrate and other HDAC inhibitors, including trichostatin A, alter p38 MAP kinase and STAT-5 signaling leading to γ-globin gene deactivation.119–121
Histone methylation
Histone methylation states can be associated with gene activation, gene silencing, or a bivalent state, to further regulate γ-globin gene expression.122–124 Protein arginine methyltransferase family member 1 (PRMT1), which regulates the action of HATs, was shown to influence γ-globin gene silencing through interactions with FoP. 125 The interaction between PRMT1 and FoP facilitates in the acetylation of Lys9/Lys14 and subsequent transcription of the adult HBB gene. 126 This suggests that PRMT1 indirectly favors activation of HBB gene expression by interacting with repressor proteins and competing for regulatory binding sites in the γ-globin promoters. Similarly PRMT5 was shown to contribute to human γ-globin gene silencing by interacting with the nuclear protein, LYAR (human homologue of mouse Ly-1 antibody reactive clone), and triggering histone H4 Arg3 symmetric demethylation by DNMT3A in adult erythroid progenitors.39,127 PRMT5 was shown to also interact with the histone lysine methyltransferase Suv4-20h1 and components of the NuRD complex to induce additional repressive epigenetic marks at the γ-globin promoter. 40 These data highlight the complexity of the relationship between histone-modifying events and recruitment of factors at the HBB locus that regulate globin gene expression.
Histone modifying agents in HbF induction
A number of HDAC inhibitors aimed at targeting epigenetic silencing of HbF expression in patients with SCD and other β-hemoglobinopathies have been investigated in clinical trials116,117, 128 (Table 1). The HDAC inhibitor, butyrate, has been studied by our laboratory119,120 and others for its role in HbF induction. 129 In 1999, Atweh et al. 117 reported that SCD patients who responded to intermittent arginine butyrate maintained high HbF levels without adverse side effects for a mean of 29.9 weeks, as long as they continued treatment 117 (Table 1). Other butyrate analogues, including phenylbutyrate, isobutyramide and other short chain fatty acids, have also been tested in clinical trials for β-thalassemia due to their capacity to induce HbF in human erythroid cells.130–132 The orally bioavailable short-chain fatty acid derivative sodium 2, 2 dimethyl butyrate (HQK-1001) was tested in the phase I/II trial consisting of 21 adult patients with β-thalassemia intermedia syndromes; however, it was not well tolerated 133 (Table 1). These findings question the potential for butyrate and its analogues as potential HbF inducer agents for treatment of patients with SCD and other β-hemoglobinopathies. Extensive efforts have been made to improve the effectiveness of HDAC inhibitors while decreasing unwanted side effects.
Epigenetic modifiers in clinical trials
A limited number of epigenetic modulators to treat SCD or other β-hemoglobinopathies have been tested in phase I or II clinical trials. Most of these drugs are intended for treatment of different forms of cancer, and as such there are major concerns about off-target effects and cytotoxicity. We summarize trials registered in Clinicaltrials.gov of epigenetic drugs that have been tested but have not proceeded to FDA approval for SCD treatment (Table 1).
MiRNAs and HbF induction
MiRNAs are short (∼22 nucleotides) noncoding RNA molecules that associate with the miRNA-induced silencing complex (mRISC) to silence gene-specific mRNA in the cytoplasm. 134 MiRNAs facilitate mRNA degradation or suppression of translation by base-pairing to complementary sequences in the 3ʹ-untranslated regions of target mRNA.135,136 The human genome encodes over 1000 miRNAs genes, which may target several different mRNAs to inhibit or restore gene expression. 137 Table 2 summarizes miRNAs that have been associated with γ-globin gene activation and HbF induction.
Summary of microRNAs and its target genes in HbF induction.
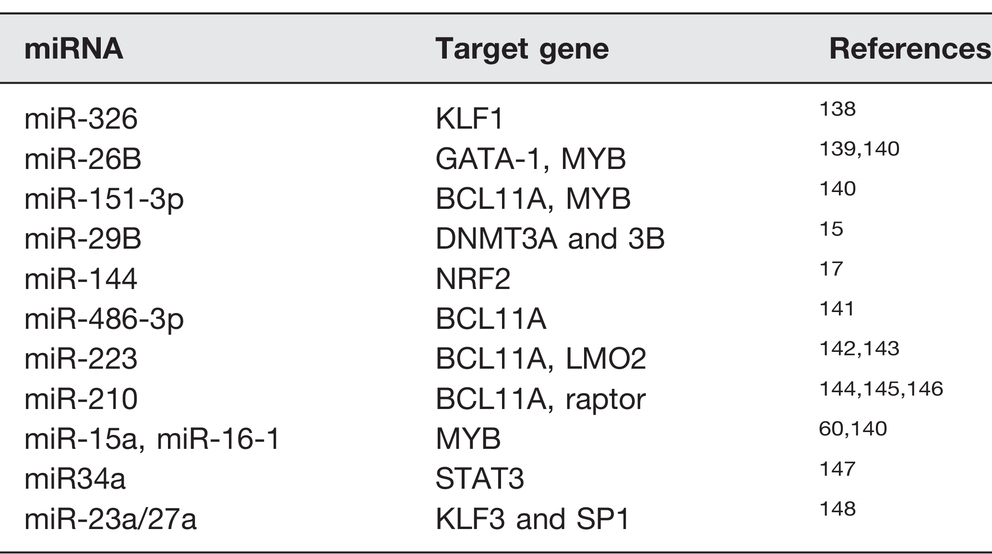
Recent reports highlight the therapeutic potential of miRNAs to ameliorate the severity of hemoglobinopathies, by also targeting globin genes or their regulatory pathways resulting in HbF induction. For instance, a study by Bianchi et al. 144 showed that miR-210 was upregulated in erythroid precursors of a thalassemia patient with high HbF levels. 144 The authors also observed that mithramycin targeted miR-210 as a mechanism of γ-globin activation in K562 cells. 144 In another study investigating SCD patients treated with HU, expression of miR-26b and miR-151-3p was associated with HbF levels at maximum tolerated doses. 149 Previous studies by our group demonstrated that miR-34a mediated HbF induction in K562 cells by silencing STAT3 expression, a known repressor of γ-globin. 147 In subsequent functional studies in normal and sickle erythroid progenitors, we demonstrated that miR-144 silenced NRF2 transcription and concomitant repression of γ-globin transcription, which was reversed in a dose-dependent manner by miR-144 antagomir. 17
This finding is important considering that we, and others, have investigated pharmacological NRF2 activators as a class of promising HbF-inducing agents for the treatment of SCD. 150 For example, Lee et al. 151 showed that overexpression of tumor promoting LIN28B decreased miR-Let7 expression and increased γ-globin gene expression and HbF levels in cultured adult primary erythroid cells, in part via targeting BCL11A expression. 151 Similarly, miR‐486‐3p mediated HbF induction in adult erythroid progenitors by inhibiting BCL11A expression although the role of direct epigenetic changes in the actions of either LIN28B or miR-486-3p remains unknown. 141 Another study demonstrated that miR-15a and miR-16–1 induced HbF by targeting the γ-globin repressor protein MYB in infants with trisomy 13, as a mechanism of delayed switching. 60
Our recently published work demonstrated that miR-29b functions as an HbF inducer in KU812 cells and normal human erythroid progenitors by targeting DNMT3 gene silencing. 15 Interestingly, overexpression of miR-29b also improves common hematological malignancies.152,153 Our published findings on the role of miR29b in HbF induction is the first study to provide evidence of a miRNA-DNA methylation interaction as a mechanism of HbF induction. Future studies to evaluate the ability of miR-29b to induce HbF in vivo are ongoing.
Non-coding small RNAs, such as miR-29b, are attractive molecules for inhibiting repressors of γ-globin gene transcription. However, there are challenges with delivery of miRNAs into cells due to their short half-life under physiological conditions and unfavorable immune response and concerns with potentially undesirable off-target effects. 154 To overcome these challenges, conjugate-mediated drug delivery has been established as a promising platform for safe and targeted small interfering RNA delivery in vivo such as cholesterol conjugation. This modification works by two primary mechanisms: (1) intercalation into the plasma membrane and internalization by endocytosis and (2) binding circulating plasma lipoproteins to promote interactions with lipoprotein receptors.155,156 Several cholesterol-conjugated small interfering RNAs have advanced to clinical evaluation such as RXI-109 for its ability to regulate connective tissue growth in age-related macular degeneration. 157 The novel concept of “miRNA supplement therapy” is currently in clinical trials for the treatment of diseases, such as myelodysplastic syndrome and cancer.158,159 It is believed that approximately 30–50% of all protein-coding genes are possibly regulated by miRNAs in health and deregulated in disease.
Summary
In summary, we highlight epigenetic mechanisms and miRNAs implicated in γ-globin gene transcription and discuss HbF-inducing epigenetic therapeutics as novel treatment approaches for SCD. Current research efforts to expand SCD treatment options support the potential ability of DNA methylation and HDAC inhibitors to be developed as HbF-inducing agents for treatment of SCD. Whether these agents play a direct role in developmentally regulated hemoglobin switching similar to major transcription repressors such as BCL11A remains a question. The discovery of the DNMT inhibitor Decitabine and HDAC inhibitor arginine butyrate for the treatment of SCD has produced sufficient clinical phenotype data and HbF induction to warrant continued investigation. However, there are many challenges associated with the development of these agents for oral administration and long-term impact on disease severity and response to treatment.160,161
Research to establish a direct role for epigenetic mechanisms and miRNAs in HbF induction is at its infancy. Additional studies to demonstrate a role in globin gene regulation and their ability to produce an additive effect on HbF induction in preclinical animal models might justify future clinical trials. The potential to target epigenetic proteins such as DNMT1, HDACs, or miRNAs involved in γ-globin gene silencing provides new therapeutic approaches to reverse globin gene silencing during adult development for the successful treatment of individuals with β-hemoglobinopathies.
Footnotes
AUTHORS’ CONTRIBUTIONS
ASD, AF, and BSP reviewed the literature and shared in writing several drafts of the paper. ASD identified and designed the review sections, figures and tables and conducted final review of paper.
DECLARATION OF CONFLICTING INTERESTS
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
FUNDING
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by HL144641-01A1 to AS-D and HL149365-01A1 to BSP.