
Abstract
Tuberous sclerosis complex (TSC) is associated with TSC1 or TSC2 gene mutations resulting in hyperactivation of the mTORC1 pathway. This mTORC1 activation is associated with abnormal tissue development and proliferation such that in the kidney there are both solid tumors and cystic lesions. This review summarizes recent advances in tuberous sclerosis complex nephrology and focuses on the genetics and cell biology of tuberous sclerosis complex renal disease, highlighting a role of extracellular vesicles and the innate immune system in disease pathogenesis.
Impact statement
Tuberous sclerosis complex renal disease has a complex mechanisms of expression. Recent findings suggest that the mechanism for tuberous sclerosis complex-associated renal cystic disease is somewhat different from that reported for associated angiomyolipomata. Tuberous sclerosis complex renal cystic disease is a cell nonautonomous trait mediated by extracellular vesicles that conscript genetically normal cells to participate to a large extent in the disease phenotype. These findings offer new insights and inroads to therapies for this rare disease.
Introduction
Tuberous sclerosis complex (TSC)-associated renal phenotypes vary considerably. These differences complicate the accurate diagnosis and thus proper treatment. Eighty percent of affected children, 1 and all adult postmortem samples 2 have renal disease, most commonly renal angiomyolipomata and cysts. Compared to the general population, 40% of TSC patients have decreased renal function even without angiomyolipomata hemorrhage or surgical interventions. 3 TSC-associated renal disease has been reported to be the most common cause of death in adults with TSC. 4 , 5 The exact number of TSC patients that go on to dialysis is unclear, in part because TSC is a rare disease and not always available as a diagnosis code for end-stage renal disease (ESRD) databases.
Genetics of TSC
TSC, designated as an autosomal dominant disorder, can impact human development and postnatally growth leading to lesions throughout the body. The birth incidence is ∼1:5800 and results in over one million affected worldwide. 6 , 7 The condition may be missed if one limits the diagnostic criteria to facial angiofibromas, developmental delay, and intractable epilepsy, as fewer than 40% of patients have these features. 8 Approximately half of TSC patients experience behavioral disorders like autism or cognitive impairment. 8
There are two gene loci associated with TSC. The TSC1 gene is on chromosome 9 and the TSC2 gene is on chromosome 16. Identification of the TSC2 gene locus was facilitated by a family with who suffered with autosomal dominant polycystic kidney disease (ADPKD) due to a translocation involving the PKD1 gene. The TSC2 gene was discovered the proband in this family had both ADPKD and TSC as they had inherited an unbalanced translocation. 9 Although both the TSC loci gene products function together to regulate the mTORC1 pathway activity, the renal disease is more severe when associated with the TSC2 gene.
Cell biology of TSC renal angiomyolipomata
Significant progress has been made focusing on the genetic mechanisms at play in TSC-associated angiomyolipomata. Angiomyolipomata belong to the PEComa family of tumors and exhibit both smooth muscle and melanocytic differentiation characteristics. 10 , 11 Histologically, angiomyolipomata contain abnormal blood vessel wall architecture surrounded by muscle-like perivascular cells and adipocyte-like cells. The interaction of the mTORC1 pathway with the microphthalmia-associated transcription factor (MITF) family helps to explain why angiomyolipomata express the melanotic marker PMEL (premelanosome protein, also known as SILV, gp100), the antigenic target for the diagnostic human melanoma black (HMB)-45 antibody. 12 This associated MITF/transcription factor E (TFE) transcription factor activity has confounded the diagnostic boundary between TSC-associated renal lesions and translocation renal cell carcinomas and PEComas. 13
Neural crest cells have been posited to give to angiomyolipomata because of the co-expression of melanocytic and smooth cell markers.14–16 This possibility has been challenged by TSC2 re-expression experiments in an angiomyolipoma cell line because this introduction changed the expression profile to resemble lymphatic endothelium rather than neural crest derived tissues. 17 A dual knock-out of Tsc2 and Cdkn2a in murine epithelial cells altered the cell expression to be somewhat similar to angiomyolipoma when injected into immunocompromised mice. 18 However, Cdkn2a mutations are found in melanoma, raising concern over the interpretation of the HMB-45 staining. Mesenchymal stem cell-like cells also have been identified in angiomyolipomata. 19 Pericytes may give to angiomyolipoma 20 and this may explain the association of TSC with vascular aneurysms that arise in angiomyolipomata, 21 aorta, 4 and brain. 22
While there are still many questions about the TSC-associated angiomyolipoma, the genetic mechanism of disease is less of an enigma. TSC-associated angiomyolipoma is thought to be phenotypically expressed by a second somatic mutation or epigenetic silencing event of the TSC non-mutant allele. A similar mechanism has be forwarded for the cystic disease in both TSC and ADPKD, though the supporting evidence for this mechanism is less robust. 23 , 24 One locus for both TSC and ADPKD is located on chromosome 16, specifically the PKD1 and the TSC2 loci. These loci account for about 70–80% of patients with ADPKD or TSC, respectively, and the disease associated with these genes is more severe than that associated with PKD2 or TSC1. The PKD1 and TSC2 genes are adjacent in a tail-to-tail orientation. This configuration is important because intron 21 of the PKD1 gene contains a polypurine•polypyrimidine tract that can adopt alternative DNA conformations. 25 This odd tract interferes with DNA replication in a polar fashion causing DNA double-strand breaks, facilitating mutagnesis. 26 , 27 Synergizing with this DNA damage, the renal osmolality inhibits DNA damage recognition,28–30 creating the “perfect storm” for potential somatic mutation.
Cell biology of TSC renal cystic disease
Many inherited renal cystic diseases are associated with the primary cilia, an organelle thought to be involved in sensing the external environment. 31 Likewise, perturbations in the mTORC1 pathway are associated with renal cystic disease, 32 , 33 and deflection of primary cilia has been shown to regulate the mTORC1 pathway. 34 , 35 Unilateral nephrectomy in transgenic mice that have undergone an “adult-induced” cilia disruption exhibited significantly higher levels of mTORC1 activity with increased cyst severity in the remaining kidney compared to the cilia-intact controls. 36
The mechanism of disease is more nuanced because although the polycystic disease-related proteins and primary cilia are mechanistically involved in polycystic kidney disease, they have a complex interaction. The loss of a polycystin protein or the primary cilia results in similar renal cystic disease phenotypes, while simultaneous inactivation of both the polycystin gene and cilia expression cause a decrease in renal cystic disease severity compared with polycystin-only knockout mice. 37 The polycystins therefore appear to modulate an unidentified signaling pathway that requires intact cilia to function, possibly implicating extracellular vesicles (EVs). EVs carry cargo including modulators of polycystic disease signaling, such as Hedgehog 38 and Wnt. 39
Tsc murine renal models exhibit elevated mTORC1 activity (phospho-S6 expression) in the cystic epithelium, but with a much lower frequency of cells that have lost Tsc gene expression.40–42 The second hit mechanism for TSC angiomyolipoma alone is incongruent with this failure to identify significant loss of heterozygosity (LOH) in the TSC cystic epithelium. 41 , 43 Cystic epithelium from patients also continue to express both tuberin and hamartin. 44 In contrast, angiomyolipomata clearly exhibit LOH and an absence of tuberin staining. 44 This low frequency of LOH is also seen in PKD1-associated ADPKD, indicating that cystic disease in general may exhibit a more complex disease mechanism. 24 , 45
EVs and TSC renal cystic disease
To understand the cystic disease in TSC better, a novel mouse model that inactivated the Tsc2 gene specifically in principal cells was generated by breeding a Tsc2fl/fl mouse to one containing Cre-recombinase driven by the aquaporin-2 promoter. The resulting mice do not have a renal phenotype at birth but acquire significant cystic disease by 19 weeks of age. 46 Curiously, although the Tsc2 gene was deleted in principal cells, the cysts were comprised of type A intercalated cells that contained an intact Tsc2 gene. 46 This is also true if the Tsc1 gene is disrupted using a similar approach. 47 Type A intercalated cells were also identified to be involved in human TSC-related cystic disease. 46 , 47
Murine lineage tracing experiments using this principal cell-targeted model of Tsc cystic disease revealed that principal cells undergo limited clonal expansion, but ultimately can provide relatively few cells to the overall cyst (Figure 1(a)). 48 The cystic kidneys contained many more interstitial EVs than non-cystic kidneys but cystic mice excreted fewer EVs in the urine than did non-affected mice. The use of mTORC1 inhibitors reduces the EV production back toward normal kidney production. 48 Cystic fluid was also rich in EVs. The loss of the Tsc2 gene caused the EV-generating cells to produce EVs that altered their effect on the transcriptome of normal renal tubular epithelium compared to EVs generated from isogenic cells with an intact Tsc gene. 48 A similar effect has been seen in an a neuronal in vivo assay, where EVs produced from Tsc1-deleted cells transformed the neighboring wild-type cells so that they became similar to Tsc1 mutant cells. 49
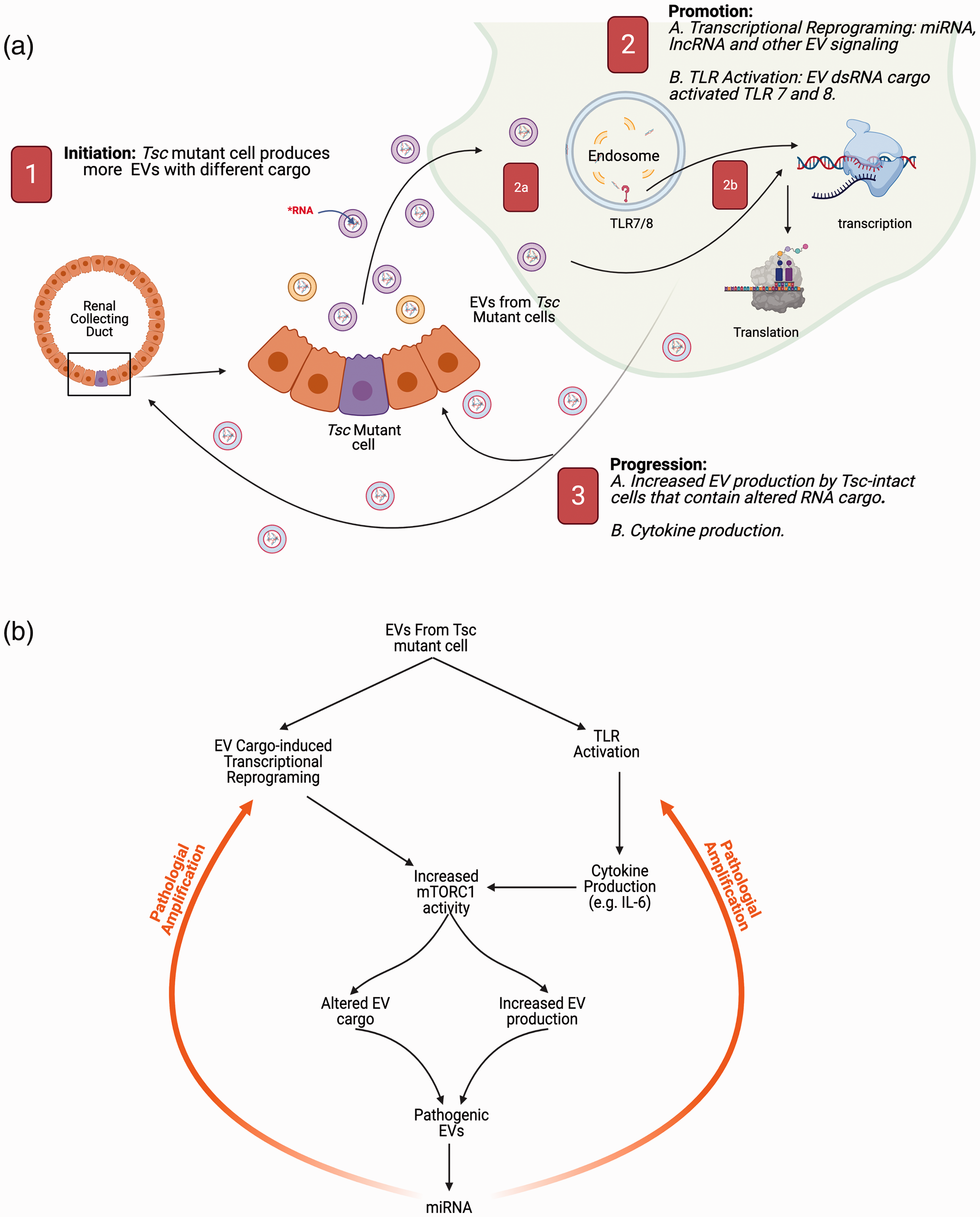
Role of EVs in renal cystogenesis. (a) Role of EVs in the initiation, promotion, and progression of TSC renal cystic disease. The cystogenic process is initiated (labeled with the white “1” in the red background) by the loss of a Tsc locus in the tubule. This genetic change results in increased EV production with different cargo. These EVs are taken up by renal tubule cells with intact TSC genes or by kidney resident macrophages. These EVs cause an increase in the mTORC1 axis and promote cystogenesis (labeled as “2”). The increase in the recipient cell mTORC1 axis is through two mechanisms. The first one (labeled as 2a) is mediated by the EV RNA cargo (miRNA and lncRNA) altering transcription in genetically intact cells, and the other mechanism involves mTORC1 activation down-stream from toll-like receptors 7 and 8 activation by the double-stranded miRNA (2b). The lesion undergoes progression (labeled as “3”) by similar mechanisms because this genetically intact cell now (labeled 3A) increases production of EVs similar to cells that have lost their TSC gene function and have an increased mTORC1 activity. This TLR activation also increases cytokine production (e.g. IL-6) which induces down-stream mTORC1 activation (labeled 3B) in adjacent cells. (b) Flow diagram of the signaling amplification network. The EVs from the mutant cells initiate a potentially pathological amplification of mTORC1 activity that drives additional miRNA and EV production that further feedback to activate the system through reprograming non-mutant cells and activating proliferation signals through TLR7 and 8 and signaling through cytokine receptors to also drive mTORC1 activity.
The role of the Tsc2 locus governing EV production is recapitulated in cell culture. Tsc2 gene deletion in mouse inner medullary collecting duct (mIMCD3) cells resulted in a more than two‐fold increase in EV production compared to isogenic cells with a non-mutant Tsc gene. 50 Proteomic analysis of EVs from isogenic cell lines except for Tsc2‐intact or Tsc2‐deleted status revealed significant protein cargo differences. 51 This raises the possibility that renal EVs that participate in tissue homeostasis may induce disease by changing the concentration of EVs or by changing their cargo. Perhaps renal compensatory hypertrophy is mediated, at least in part, by EVs. In the cystic disease state, the EVs produced by renal cyst epithelia may drive growth contributing to disease. Such EV reprograming of adjacent genetical intact cells involve miRNA EV cargo, because in cell culture experiments, quantitative polymerase chain reaction (PCR) analysis revealed the downregulation of miR-212a-3p and miR-99a-5p in EVs derived from Tsc2 mutant cells compared to EVs purified from Tsc1 mutant isogenic cells. These specific EV-derived miRNAs are expected to impact the mTORC1 activity in the recipient cell by altering the transcriptome and impacting the proliferative capacity. 52
Physiological and pathological roles of EVs in the kidney
The intercellular communication of EVs has a larger role in renal homeostasis and there also may be a role in the collecting duct plasticity. Embryologically, collecting duct epithelium arises from the ureteric bud and differentiates into principal and intercalated cells. 53 These epithelial cells display significant adaptability, and intercalated cells can participate with the principal cells in regulating the intravascular volume by way of tubular NaCl resportion. 54 In addition, type A and type B intercalated cells can interconvert between the two subtypes to help control systemic acid–base balance. 55 , 56 Both intercalated cell subtypes use carbonic anhydrase II, and knocking out the CA2 gene results in intercalated cell population depletion with a proportional increase in the principal cell population. 57 Cortical collecting duct cell plasticity also includes the relative ratio of intercalated cells to principal cells. 58 , 59 For example, potassium homeostasis is facilitated by principal cell secretion and intercalated cell absorption. Cell lineage tracing experiments in the context of potassium depletion demonstrated intercalated–principal cell interconversion in the medullary collecting duct. 60 Likewise, lithium can increase the percentage of intercalated cells through Notch and Foxi1 pathway activity, helping to explain lithium-associated diabetes insipidus. 58 ,61–63 Beyond plasticity, EVs also may help explain the disparate finding of collecting duct epithelial cell renin in the context of little or absent corresponding mRNA. 64 , 65 Such protein could be EV cargo from the vascular compartment or more upstream tubular epithelial cells.
EVs can mediate cellular proliferation and altered cellular function for normal organ homeostasis, but perhaps they are even more critical in response to disease states. When cells are stressed, they most often increase their EV production in addition to altering the EV cargo. This is true in human disease. Diabetic vascular dysfunction is mediated by transcriptionally modifying RNAs, 66 and more importantly to EV RNA cargo. 67 During normal physiological homeostasis, cells synchronize their metabolic activity, gene expression, and basic cellular processes. When cells are challenged or stressed, for example by a reduction of nutrients, they communicate to adapt their metabolism to the new condition. This same EV communication system can effectively conscript genetically normal cells to adopt a TSC disease phenotype 48 (Figure 1(a)). Once taken up, the EV cargo RNA can epigenetically alter recipient cell activity in two ways. First, the miRNA and lncRNA can alter cellular transcriptional activity and effectively reprogram the cellular activity. The second mechanism actually may activate an immune/inflammatory pathway. 68 , 69
Dendritic cells help regulate innate and adaptive immunity in nonlymphoid tissues, and tissue-resident dendritic cells participate in an extensive, contiguous network in the kidney and appear to be positioned to respond to environment stimuli. 70 This immune cell network also functions as important facilitators in renal cystogenesis. Renal resident macrophages phenotypically change from R2b to R2a during postnatal maturation, coincident with the timing when cyst formation slows considerably in genetic models of renal cystogenesis. Renal injury increases the number of juvenile-like resident macrophages in cilia mutant mice and accelerates cyst growth. Using a CSF1R kinase inhibitor in these animals to alter the resident macrophage activity assuaged cystogenesis. 71 There is innate immune functional overlap between renal tubular cells and the resident macrophage population that may contribute to this process in cystogenesis (Figure 1(a)). 72
Environmental factors can induce EVs that impact immune cell fate decisions, activation, polarization, and memory cell development important to immune homeostasis. 73 , 74 This effect involves pattern recognition receptors (PRR), such as the toll-like receptors (TLR), that recognize pathogen characteristics to induce an immune response including cytokine and reactive oxygen species secretion. 75 Under normal circumstances, such PRR activation initiate immune cell recruitment and tissue repair. Pathological conditions of sustained PRR-signaling can result in autoimmunity or lethality. 76 , 77 This miRNA activation of the renal tubular epithelium, with or without the tissue resident mononuclear cell involvement, may further activate the mTORC1 signaling pathway through the TLR7 and 8 (Figure 1(b)). Such pathological amplification of mTORC1 activation through both mechanisms could go a long way to help explain the “phenotypic spreading” described in Tsc mutant cell systems. 49
This sort of cell nonautonomous disease trait is not new, and a role for EVs in cellular reprogramming has been observed in a targeted mouse model that inactivated Brca1. Chodankar et al. use a targeted Brca1 inactivation in mouse ovarian granulosa cells. The inactivation of the Brca1 gene in granulosa cells induced cystic tumors with genetically intact Brca1 genes in the ovaries and uterine horns. The authors hypothesized that the loss of Brca1 by granulosa cells caused them to secrete something that caused the tumor to arise in a different cell type 77 , and we posit the mutant granulosa cells secreted EVs that lead to the tumors to arise in the uterine horns.
Key questions and future directions
There are several initial key questions that will direct future investigations. The first question deals with the role of EVs in coordinating growth in developing tissue, and healing in existing, but damaged renal tissue. The renal cystic disease finding may have broader biological impact in general, and these findings then may offer a new insight into other proliferative diseases. Another key question focuses on how this signaling is directed and whether there is an addressing or targeting system involved in EV trafficking. If such an EV targeting system is at play, then this may offer a way to alter the EV signaling and possibly offer insights into therapy. Likewise, another key question focuses on the role of the cellular response to the double-stranded RNA found in the EV cargo. Separate from the noncoding RNA effects on altering transcription, this effect, for example through the PRR, would also provide additional therapeutic avenues. Taken together, these questions and directions have great potential for leveraging the biology to reduce the disease burden.
Conclusions
While TSC renal disease is a leading cause of morbidity and mortality, new insights into the disease pathogenesis also offers new therapeutic avenues. Understanding the detailed contributions may offer additional pathways to modulate the signaling pathway that can be leveraged to improve patient outcome. Beyond TSC, EVs and the innate immune system may be at play in other renal proliferative diseases such as other forms of renal cystic disease and malignancy. While further research is required to better illuminate the disease mechanisms and therapeutic potential, there is great potential for benefit.
Footnotes
AUTHORS’ CONTRIBUTIONS
Conceptualization: FZ, JJB, PK, and YY; Resources: JJB; Writing—original draft preparation: FZ, JJB, PK, and YY; Writing—review and editing: FZ, JJB, and PK; Supervision: JJB; Funding acquisition: JJB.
ACKNOWLEDGEMENTS
I (the corresponding author) am grateful for this opportunity to publish a paper in remembrance of Peter Stambrook, PhD. Having the privilege of his friendship was one of the best things I carry with me from my time in Cincinnati. The figure was created with BioRender.com.
DECLARATION OF CONFLICTING INTERESTS
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
FUNDING
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the DoD grant W81XWH-14-1-0343 (JJB), Federal Express Chair of Excellence (JJB), and Children’s Foundation Research Institute (JJB).