
Abstract
Rare diseases affect nearly 300 million people globally with most patients aged five or less. Traditional diagnostic approaches have provided much of the diagnosis; however, there are limitations. For instance, simply inadequate and untimely diagnosis adversely affects both the patient and their families. This review advocates the use of whole genome sequencing in clinical settings for diagnosis of rare genetic diseases by showcasing five case studies. These examples specifically describe the utilization of whole genome sequencing, which helped in providing relief to patients via correct diagnosis followed by use of precision medicine.
Keywords
Impact statement
Rare diseases affect nearly 300 million people globally with most patients aged five or less. Traditional diagnostic approaches have provided much of the diagnosis; however, there are limitations. For instance, simply inadequate and untimely diagnosis adversely affects both the patient and their families. This review is very important in the current time because the sequencing technologies are rapidly changing and the use of WGS as a diagnostic test is becoming more practical and feasible to solve the increasing number of undiagnosed rare diseases. This review differentiates current sequencing schemes concerning their cost per sample, types of variants detected, sequencing depth along with its pros and cons. Additionally, it advocates the use of WGS in clinical settings for the diagnosis of rare genetic diseases by showcasing five case studies where utilizing the technique has helped in providing relief to patients via correct diagnosis followed by the use of precision medicine.
Introduction
A genetic disorder is defined as an abnormality caused by either single (monogenic), multiple (polygenic) gene mutations or chromosomal abnormalities. Monogenic disorders are mostly Mendelian in nature. They usually arise during the development of the fetus, making them visible at birth and are diagnosed based on family history. Regretfully, most monogenic disorders last specific treatment. In contrast, polygenic mutations are multifactorial in nature, showing their signs and symptoms due to a combined effect of multiple polymorphic genes in combination with external environmental factors. Diseases that occur due to polygenic mutations are called “complex” diseases. They are non-Mendelian in nature, show reduced penetrance, yet occur more frequently than single-gene disorders. Here, the effect is more gradual, with disease symptoms appearing at a later stage of life. The most significant difference between single and complex gene disorders pertains to the degree to which genetic mutations alter the phenotype.
Rare and common diseases are defined as “rare” or “common” based on their relative prevalence (Figure 1). Rare diseases affect nearly 300 million people worldwide. 1 They vary significantly across different parts of the world each with different mutations, phenotype, and diagnostic methods (Table S1). As per Orphanet, as of 2021, there are about 7000 rare diseases with genetic causes, leading to nearly 80% of all cases. Regretfully, symptoms are often misrepresented leading to incorrect diagnosis and delay in therapy. Moreover, rare diseases are often severe with most of them incurable. As patients affected by rare diseases are few, research in disease diagnostics and therapeutics has not reached its true potential, rendering immense suffering to the patient and their families. Nevertheless, accurate and timely diagnosis is necessary because it helps physicians manage their patients as well as counsel their families.2,3 Hence, this work focuses on “rare genetic diseases.”

Worldwide prevalence of rare and common disease per 100,000 people. Most rare diseases have low prevalence varying from 10 to 50 per 100,000. On the other hand, common diseases occur more frequently, ranging from 50 to 10,000 per 100,000. (Data adapted from January 2020 Orphanet and World Health Organization (WHO) reports.)
In 2011, the International Rare Diseases Research Consortium (IRDRC) started with the aim to provide accurate diagnosis and suitable therapy to rare diseases in the shortest possible time.4,5 According to IRDRC, since 2010 more than 800 novel rare diseases have been reported with close to 4000 associated genes. As rare genetic diseases are not easily identified on phenotypes, determining the exact mutation causing the genetic disease is necessary. Traditional genetic diagnosis involves both conventional screenings such as chromosomal microarray (CMA) as well as screening entire exomes and genomes to determine the exact cause of the disease. 6 Hence, this review advocates the use of whole-genome sequencing (WGS) for diagnosing rare genetic diseases; enumerates both traditional and WGS based screening frameworks; specifies six case studies where WGS successfully identified rare genetic diseases which were previously undetected via conventional sequencing; and finally highlights the importance of WGS as a first-tier test with a caution on potential hurdles that need to be resolved before bringing it fully into a clinic setting.
Traditional genetic screening
First introduced in the 19th century, conventional genetic screening starts with G-banded karyotyping which helped in identifying chromosomal abnormalities in number, translocations, inversions, or amplifications of chromosomal segments. The process starts by treating metaphase chromosomes with trypsin enzyme causing the chromatin structure to relax. Thereafter, mitotic cells arrested in the metaphase stage of the cell cycle are stained with Giemsa dye producing between 400 and 800 different bands (G-banding) distributed across 23 pairs of human chromosomes. The banding pattern that is numbered on each arm of the chromosome from centromere to telomere is easily identified and any structural chromosomal changes are described accordingly. Examples of its diagnostic capability include showing trisomy 21 leading to Down syndrome and an extra X-chromosome causing Klinefelter syndrome.7,8 However, karyotyping is limited in its scope because it is unable to detect chromosomal changes smaller than three million base pairs (Mb). 9
First introduced in 1935, a technique called fluorescence in situ hybridization (FISH) was introduced showing better sensitivity as compared with its predecessor karyotyping.10,11 Its applications included prenatal screening to detect aneuploidy, suspected malignancies, gene rearrangements, and deletions close to telomeres (as in the case of leukemia). However, it too had limitations in its diagnostic capacity, as FISH exhibited low resolution (300 kb) noticing only those chromosomal locations that were specifically targeted by FISH probes. 12
Introduced in 1993, chromosome microarray analysis (CMA) replaced both FISH and karyotyping, as it enabled the detection of submicroscopic variations not detected by conventional techniques. The principal behind CMA involves the isolation of genomic DNA of both healthy control and a diseased individual. The two genomes are enzymatically broken down, differentially labeled with different fluorochromes, and co-hybridized on a microscopic glass slide to which cloned DNA segments from a representative genome are immobilized. 9 Copy number variations (CNVs) along the length of chromosomes are detected by measuring the differences in fluorescence signals and normalized to compare data between patient and control samples. 13
CMA facilitates the diagnosis of novel rare diseases, as it detects CNVs particularly in neonates suffering from congenital birth defects. 14 Nevertheless, CMA is unable to detect small chromosomal rearrangements and somatic mosaicism. Therefore, with limitations still unaddressed, conventional gene discovery necessitated the movement towards next-generation sequencing for diagnosing rare genetic diseases as highlighted in Table 1.
Genetic screening tests while diagnosing rare disease.

Note: Genetic screening tests ranges from analyzing chromosome via light microscope to detecting copy number variation to detecting specific coding regions to the full genome. With increase in resolution, the number of variants detected also increases. WGS detects the highest number of variants by covering the entire genome showcasing the largest diagnostic yield making it an ideal technique for detecting variants left undiscovered from traditional techniques.
NGS-based screening
Next-generation sequencing (NGS) uses high throughput sequencing technologies to sequence (i) coding regions of targeted genes to (ii) entire exomes and (iii) genomes. The general steps for NGS analysis are depicted in Figure 2. With rapidly decreasing sequencing cost and the advent of long-read sequencing, genomic medicine has allowed clinicians to devise new strategies for prevention, diagnosis, and therapy of rare genetic diseases. 15 The first report of using NGS in diagnosing a rare disorder called Freeman-Sheldon syndrome came in 2009 by identifying the MYH3 gene as a causative agent, 16 followed closely by identifying disease-causing genes for both Miller syndrome 17 and Kabuki syndrome, 18 both of which were not possible via conventional screening methods.
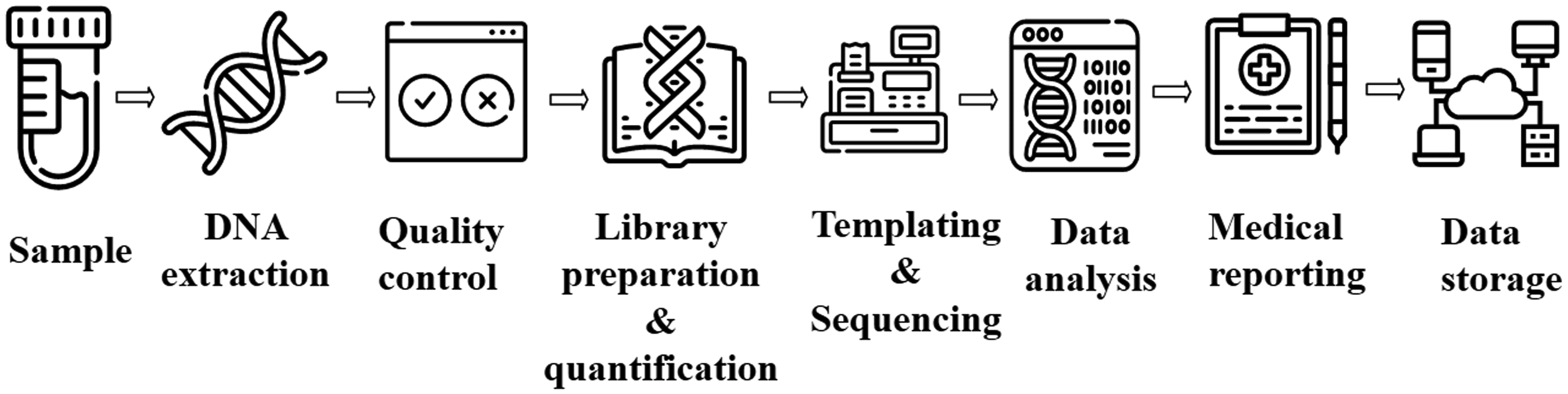
General NGS Workflow starting from sample collection till data storage. The process starts from DNA extraction from samples followed by quality control, library preparation, and subsequent sequencing by a sequencing machine. If the sequencing process completes successfully, bioinformaticians conduct appropriate analysis as per need.
NGS has played a pivotal role in identifying more than 180 pathogenic mutations, 19 including heterozygous mutations where only a single copy of mutant is present in homologous chromosome. 20 NGS-based screening methods include (i) targeted, (ii) whole-exome, and (iii) whole-genome sequencing. 21
Targeted gene panels
Targeted sequencing is performed by either hybridization-based targeted enrichment or PCR-based amplicon sequencing. It is favored because (i) it provides quick results because it screens only known pathogenic variants to known disease gene, (ii) can detect rare genetic variants at high sequencing depth (
WES
WES covers the entire coding region (exome), which makes up to 2% of the genome. The process involves enrichment of coding regions of the genome, regulatory regions, and other functionally annotated regions of interest such as miRNA. It has been successfully employed to identify genetic causes for neurological disorders, 23 intellectual disability (ID), 24 and autism spectrum disorders 25 to name a few. It is popular because of its (i) low cost, (ii) relative abundance of pathogenic mutations in protein-coding regions, (iii) easy data storage, and (iv) processing. However, as WES only targets the exome (2% of the genome), it is unable to capture pathogenic variations that occur in the remaining 98% leaving us with WGS to look forward to.
WGS
By sequencing the entire genome, WGS can potentially detect all pathogenic variations. Gradually, WGS is becoming an effective first-tier test in cases where physicians face diagnostic ambiguity. General steps for WGS remain the same as that of WES as shown in Figure 3. When compared, WGS outperforms WES with similar coverage, as WGS (i) is less sensitive to GC content; (ii) provides more uniform coverage; (iii) capable of identifying both exome and non-coding pathogenic variants; (iv) suited to detect SNPs, CNVs, inversions, indels (in case of small read WGS), whereas long read WGS can recognize chromosomal rearrangements like tandem repeats31–34; (v (effective in trio-based screening35,36; (vi) proficient in detecting long repetitive regions (as in the case of Oxford Nanopore and PacBio) helping to diagnose tandem-repeat diseases, (vii) determine structural changes and transposable elements (TE) insertions (in case of Oxford Nanopore and PacBio)37–42; and (viii) senses SNVs and large-scale deletions in mitochondrial genome-causing disorders43,44 like Kearns-Sayre syndrome, Pearson’s syndrome, 45 and Addison disease. 46
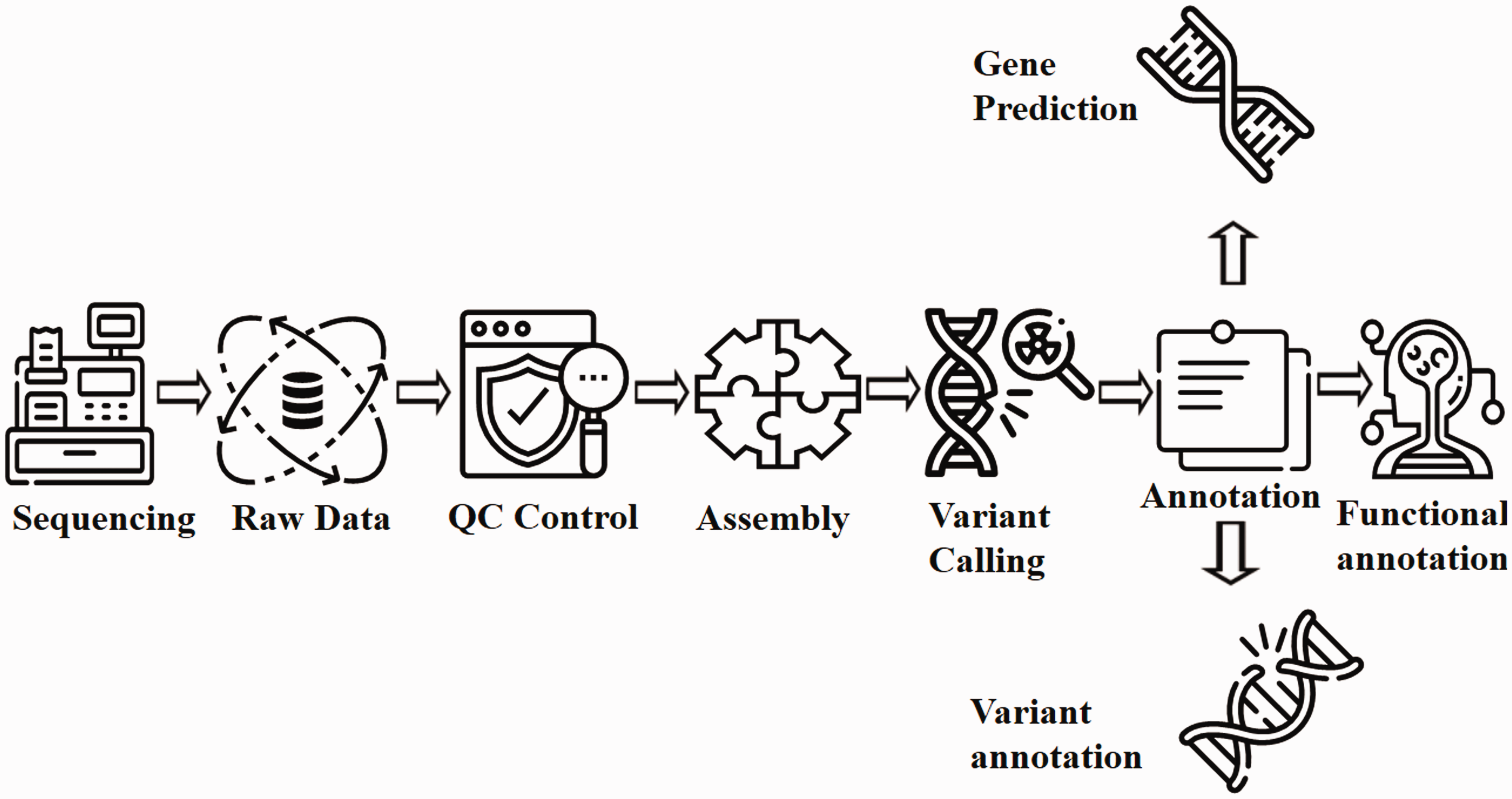
Flow chart for WGS analysis pipeline. Raw data generated from the sequencing process undergoes extensive cleaning and quality control. Thereafter, the filtered reads are joined together via de-novo or comparative assembly to form contiguous sequences (contigs). Contigs are connected via scaffolding to obtain draft assemblies. Thereafter, the assembled genome is searched for variants and annotated for identifying gene locations, determining the function of those genes and quantifying the impact of variation on proteins. Readers may employ either Genobuntu (Abbas WA, Genobuntu Package for Next Generation Sequencing. http://sourceforge.net/projects/genobuntu/) or Baari, both providing sufficient tools and software for the entire analysis pipeline.26–30
Figure 3 summarizes the series of interconnected analyses referred to as “pipeline” in the WGS process. Whereas Table 2 outlines the merits of WGS compared with WES, and Table 3 delineates some case studies where WGS proved more effective than WES and conventional genetic screening.
Comparison of targeted gene sequencing, WES, and WGS.
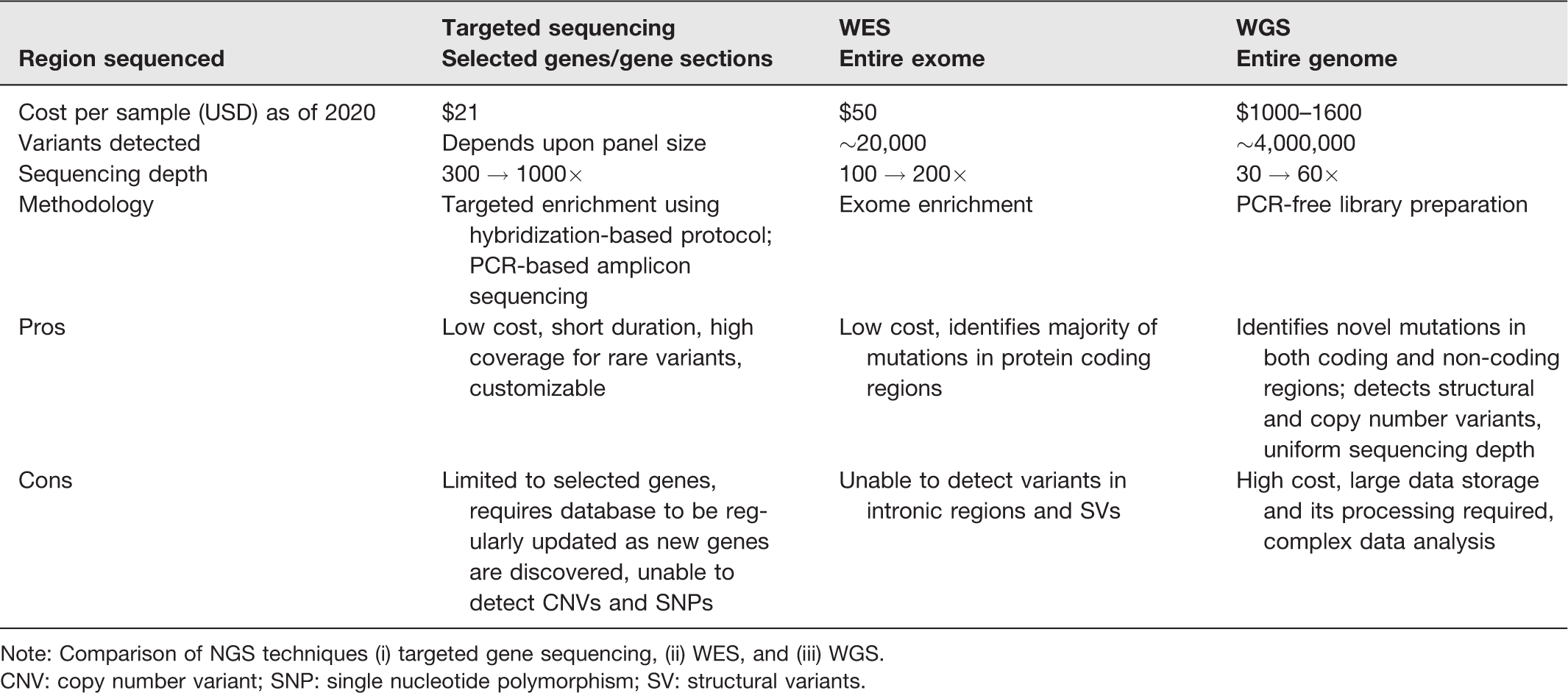
Note: Comparison of NGS techniques (i) targeted gene sequencing, (ii) WES, and (iii) WGS.
CNV: copy number variant; SNP: single nucleotide polymorphism; SV: structural variants.
List of case studies where WGS was used as a diagnostic test.
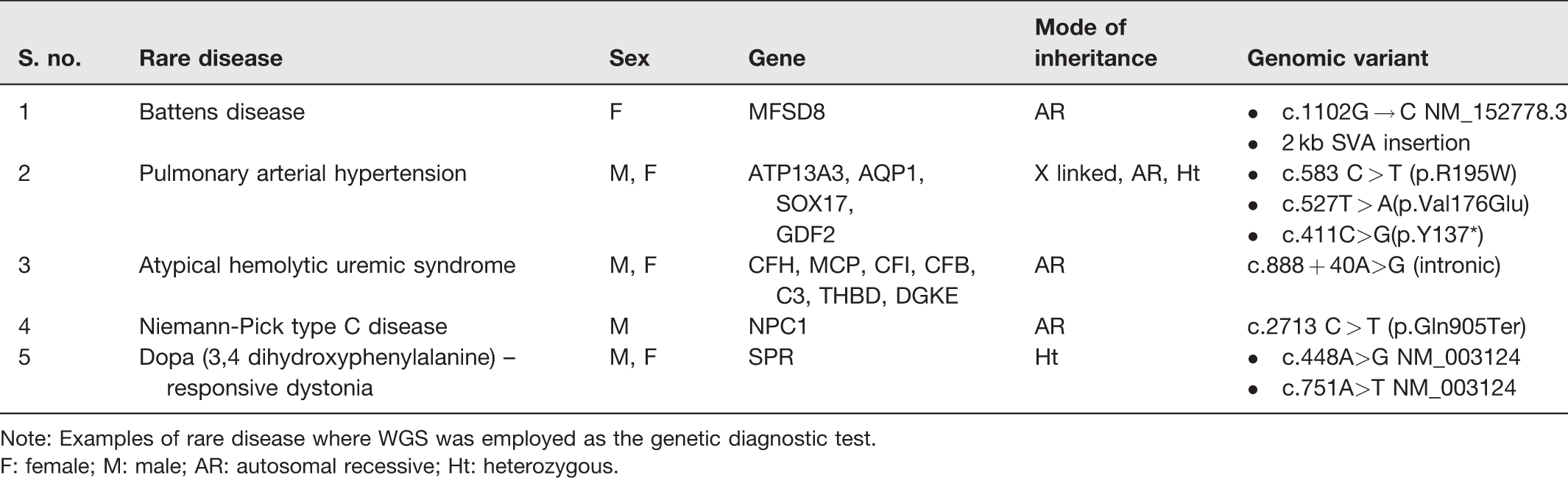
Note: Examples of rare disease where WGS was employed as the genetic diagnostic test.
F: female; M: male; AR: autosomal recessive; Ht: heterozygous.
Diagnosis of some rare diseases based on WGS
Batten’s disease
Batten’s disease, also called Juvenile Neuronal Ceroid Lipofuscinosis, is primarily caused by a mutation in the CLN3 gene. 47 Batten’s disease has an autosomal recessive mode of inheritance with initial symptoms that include sudden onset of blindness, ataxia, dysarthria, dysphagia, and seizures.
In a case study, magnetic resonance imaging (MRI) images of a six-year-old patient's head revealed cerebral and cerebellar atrophy, whereas skin biopsy showed an irregular pattern of lysosomal inclusions. Targeted gene panel revealed heterozygous single known pathogenic mutation in MFSD8 gene with no other mutation. Thereafter, medical experts conducted a trio-based WGS screening to reveal a group of 2 kb SVA (SINE-VNTR-Alu) insertion in MFSD8 intronic region located in both the patient and her mother, thereby changing MSD8 splicing and translation effect. 48 This successful diagnosis via WGS helped develop Milasen (a 22-nucleotide antisense oligonucleotide) as personalized antisense oligonucleotide therapy49,50 with its repeated injections for one year resulted in improvement of patient’s condition by reducing the number of seizures by more than 50%. 51
Pulmonary arterial hypertension
Hereditary pulmonary arterial hypertension (PAH) is a rare disorder characterized by blockage of arterioles in the lung, leading to pulmonary vascular resistance. 52 Earlier, PAH was assumably caused by an injury to smooth blood vessels of the lung. However, this alone could not account for 15–20% of inherited cases of PAH. Later, a study in 2010 found a mutation in BMPR2 gene. 53 Still, some blanks needed filling. It was only when researchers applied WGS that four additional causative genetic variations for PAH were found in ATP13A3, AQP1, SOX17, and GDF2 genes. 54
Atypical hemolytic uremic syndrome
Atypical hemolytic uremic syndrome (aHUS) is a rare disorder characterized by features of thrombocytopenia, non-immune microangiopathic hemolytic anemia, and acute renal failure. 55 Diagnosing aHUS, without a family history, is difficult, as the exact cause of genetic alteration remains unidentified. WES analysis discovered mutations in at least seven genes with CFH gene being termed the most dominant factor linked to aHUS.
A study inducted two unrelated families and conducted both WES and WGS screening. While WES was unable to link any significant mutation in previously known genes, WGS was able to determine a non-coding mutation (c.888 + 40A>G) in DGKE gene resulting in a disrupted form of DGKE mRNA, thereby adversely affecting protein catalytic sites. These WGS-driven results had direct implications on clinical management of the disease as physicians stopped administering both plasma therapy and eculizumab (a drug commonly used to treat aHUS), as both seemed to have no link with the causative agent DGKE gene.
Niemann-Pick type C disease
Niemann-Pick type C disease (NPC) is a rare autosomal recessive disorder, 56 characterized by intracellular cholesterol trafficking, neurological disorder, reduction in bile flow and liver abnormalities due to lipid accumulation within liver cells (specifically, hepatocytes). Previously, mutations in NPC1 gene addressed up to 95% of the affected families. 57 This was also confirmed in a case study where an infant (male) showed features indicative of NPC, even though the child’s parents were not cousins. After liver biopsy and electron microscopy, it was only WGS that confirmed the mutation in NPC1 gene as the causative agent of NPC disease. 58 This enabled the physicians to employ appropriate therapies to (i) delay neurodegeneration and (ii) prevent irreversible damage to the patient’s neurons. 59
Dopa (3,4-dihydroxyphenylalanine) responsive dystonia
Dopa (3,4-dihydroxyphenylalanine) responsive dystonia (DRD), also known as Segawa syndrome, is a heterogeneous rare inherited movement disorder, 60 where the patient's lower limb muscles contract uncontrollably. Patients with DRD lack enzymes involved in dopamine syntheses like GTP cyclohydrolase 1 (GTP-CH-I) or sepiapterin reductase. Previous studies on DRD showed an autosomal dominant mutation in GCH1 gene as the leading cause of DRD. 61
However, a study that investigated the entire genome of fraternal twins diagnosed with DRD, revealed heterozygous mutations in the SPR gene. This mutation reduces the synthesis of tetrahydrobiopterin, an essential cofactor required for the synthesis of dopamine and serotonin. This WGS-driven finding had immediate implications on clinical therapy, as physicians administered L-dopa therapy to both fraternal twins. The therapy helped improve movement coordination, enhance sleep and focus, boost exercise capability, and reduce the frequency of laryngeal spasms. 62
Application of WGS on national healthcare systems
As an important milestone, WGS has shown significant potential when applied to large cohort studies involving both Swedish and UK’s healthcare systems. For instance, the Karolinska Institutet in Sweden conducted an extensive study involving 4437 patients under the “clinical academic” collaborative model using WGS. This clinical–academic model proved promising, as their collaboration resulted in determining the cause of rare genetic diseases in nearly 1200 patients of which 54% (∼650 patients) were previously undiagnosed using previous diagnostic frameworks. 63
In another study, the UK's healthcare system applied WGS to 13,037 cohort participants, of whom 9802 were diagnosed to have rare diseases. Out of the 9802 patients, WGS was able to determine the genetic causes of 1138 patients showing the effectiveness of the framework concerning rare genetic diseases. 64
Conclusions
Rare diseases are chronic and often life-threatening, hence requiring accurate and timely diagnosis both for disease management and personalized therapy. The review advocates employing WGS as a first-tier genetic screening test for rare genetic diseases. With the increase in unsolved cases following WES, more disease-associated genes and variants remain to be explored. This is because most of the knowledge about disease-causing variants revolves around the coding region but less is known about the role of non-coding and structural variants. This calls for alternate approaches such as sequencing the entire genome, third-generation long-read sequencing, and transcriptome sequencing. Clinical WGS promises to deliver its potential in disease management, accurate diagnosis, and solving unknown cases which remains a burden for both patients and healthcare workers. WGS could hugely impact pediatric genomics as a study diagnosed rare genetic diseases in two critically ill newly born children within 50 h of WGS screening as a first-tier test. 65
Clinical WGS could pave the way for designing personalized therapy for the patient and providing enough information for genetic counselors to guide affected families regarding the risks of genetic mutations running through generations. Due to rapidly decreasing sequencing costs, WGS is becoming more accessible and an important genetic screening test for rare diseases.
Despite its potential, some hurdles need to be solved. This includes (i) the availability of powerful computing systems with (ii) appropriate bioinformatics programs coupled with (iii) technical personnel that can read and interpret data from a clinical standpoint. Moreover, as the data is huge, (iv) storage and transfer of raw data files is challenging, and although regulatory bodies like the American College of Medical Genetics and Genomics have published guidelines for employing WGS in clinical settings, (vi) individual interpretation is still quite varied.
To address some of these challenges, the Medical Genome Initiative was formed with the (initial) goal to publish recommended clinical and laboratory practices for applying WGS into medicine. 66 It is important to develop, constantly update, and maintain a detailed rare mutation database to facilitate diagnostic/prognostic studies across different parts of the world. Moreover, it is imperative to study epigenetics, transcriptome, proteome, and functional analysis of the genome for an improved understanding of the disease mechanism for us to devise targeted therapies. Nevertheless, with rapidly decreasing sequencing costs and an intensely collaborative approach, WGS is expected to become a standard first-tier approach for diagnosing rare genetic diseases.
Footnotes
AUTHORS’ CONTRIBUTIONS
HN participated in the design, interpretation and writing of the manuscript; BW supervised the entire study, including writing, proof-reading and editing the article; SS assisted in developing figures; FA highlighted clinical application; IW, AK, US and SS helped edit the manuscript and provided valuable suggestions. All authors have read and agreed to the submitted version of the article.
DECLARATION OF CONFLICTING INTERESTS
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
FUNDING
The paper has been partly supported by Sabz-Qalam, Grant # SQ-2019-Bioinfo-1.