
Abstract
Biological therapies against breast cancer patients with tumors positive for the estrogen and progesterone hormone receptors and Her2 amplification have greatly improved their survival. However, to date, there are no effective biological therapies against breast cancers that lack these three receptors or triple-negative breast cancers (TNBC). TNBC correlates with poor survival, in part because they relapse following chemo- and radio-therapies. TNBC is intrinsically aggressive since they have high mitotic indexes and tend to metastasize to the central nervous system. TNBCs are more likely to display centrosome amplification, an abnormal phenotype that results in defective mitotic spindles and abnormal cytokinesis, which culminate in aneuploidy and chromosome instability (known causes of tumor initiation and chemo-resistance). Besides their known role in cell cycle control, mitotic kinases have been also studied in different types of cancer including breast, especially in the context of epithelial-to-mesenchymal transition (EMT). EMT is a cellular process characterized by the loss of cell polarity, reorganization of the cytoskeleton, and signaling reprogramming (upregulation of mesenchymal genes and downregulation of epithelial genes). Previously, we and others have shown the effects of mitotic kinases like Nek2 and Mps1 (TTK) on EMT. In this review, we focus on Aurora A, Aurora B, Bub1, and highly expressed in cancer (Hec1) as novel targets for therapeutic interventions in breast cancer and their effects on EMT. We highlight the established relationships and interactions of these and other mitotic kinases, clinical trial studies involving mitotic kinases, and the importance that represents to develop drugs against these proteins as potential targets in the primary care therapy for TNBC.
Keywords
Impact statement
Some mitotic kinases regulate key cellular processes, including the centrosome cycle, and the regulation of the cell cycle phases: mitosis and cytokinesis. Moreover, several of these kinases have a clear role in cancer progression and some inhibitors against these kinases have had promising results in clinical trials. Unfortunately, to date, there are no current clinical trials with inhibitors against some of these proteins, including Hec1 or Nek2. Thus, in this work, we put into perspective the interconnections between several mitotic kinases that control the EMT process, ongoing clinical trials against these kinases, and the imminent importance to develop drugs that target them to improve and diversify directed breast cancer therapies in a near future.
Introduction
Breast cancer is a heterogeneous disease that is classified into molecular subtypes according to their progesterone (PR) and estrogen (ER) hormone receptors (HR) and Her2 amplification status, and by mRNA expression profiling.1,2 There are five major molecular (pathological) subtypes of breast cancer: Luminal A (ER+PR+Her2–), Luminal B (ER+PR+ and Her2 + or –), Her2 enriched (ER-PR-Her2+), normal-like, and triple-negative breast cancer (TNBC or ER-PR-Her2–). TNBC associates with a high proliferative index, distant metastases, and poor survival.3,4 TNBC has higher rates of centrosome amplification-driven aneuploidy and chromosome instability (CA/CIN), which are phenotypes that associate with tumor initiation, invasion, and resistance to chemotherapeutic agents used in TNBC, such as taxanes, relative to other subtypes.5–10 Previous studies have reported that breast cancer HR status is an important predictor of survival among different ethnic groups. For example, Black women are more likely to be diagnosed with aggressive, HR negative subtypes, including TNBC.11,12 Despite the availability of pharmacological treatments for Luminal and Her2+ tumors, current directed therapies offer no significant effectivity against TNBC. Therefore, research into potential biological targets to treat this poor prognosis breast cancer subtype is of crucial relevance since TNBC is more prone to develop chemo-resistance and radio-resistance compared with the other molecular subtypes. 13
Notably, many TNBCs have an active epithelial-to-mesenchymal transition (EMT) program that confers metastatic properties to cancer cells through enhancement of migration, invasion, and metastasis. 14 EMT is a hallmark of cancer and involves a complex network of genes that mediate physiological and molecular changes in cell morphology, basal-apical polarity, and epithelial to mesenchymal phenotype. 14 EMT is characterized by the loss of cellular epithelial junctions and the apical-basal polarity that leads to the reorganization of the cytoskeleton and morphological changes.14,15 These cellular processes are regulated by EMT transcription factor families such as SNAIL (SNAI 1–3), bHLH (Twist1 and Twist2), and ZEB (Zeb1 and Zeb2). 16 These transcription factors repress the expression of mRNAs coding for epithelial genes, such as E-cadherin, Claudin, Occludin, Desmoplakin, Cytokeratins, and ZO-1, among many others. 14 Meanwhile, the SNAIL, bHLH, and ZEB families activate mesenchymal genes, such as fibronectin, N-cadherin, matrix metalloproteases (MMPs), among several others. An important event that occurs during EMT is the loss of E-cadherin, a process known as cadherin switch, where E-cadherin is replaced by another cadherin. The cadherin switch is context-dependent, and although a switch between E and N cadherins is well known, E-cadherin may be replaced by other cadherin types. 17 In addition to these molecular changes, EMT regulation also occurs at the microRNA, long noncoding RNA, chromatin, and post-transcriptional levels. 18
Ultimately, the acquisition of mesenchymal features allows the cells to invade and migrate to distant sites forming secondary tumors. 14 EMT correlates with cancer stem cells and may facilitate the generation of cancer cells with mesenchymal phenotype needed for dissemination as well as the self-renewal properties needed for initiating secondary tumors. EMT may facilitate metastasis through the Tumor growth factor Beta (TGF-β) signaling 19 and the activation of β-catenin and AKT pathways by Twist1. 20
EMT used to be described as a binary process where cells displayed an epithelial or mesenchymal phenotype. However, recent studies demonstrated that EMT occurs in gradual (hybrid) steps and each step is characterized by distinct morphologies, gene expression, and varying capacity for proliferation, invasion, plasticity, and invasiveness.14,21–26 The hybrid EMT state may help the metastasis process since once the cells reach a distant site they need to revert to an epithelial phenotype to form the tumor, this process is referred to as MET (mesenchymal-to-epithelial transition). In addition to the role of EMT to facilitate metastasis, EMT may contribute to drug resistance and immunosuppression. 27
Despite the research advancements to elucidate the molecular signaling underlining EMT in TNBC and other cancers, the exact mechanisms remain unknown. Recently, we found that overexpressing any of the three E2F activators (E2F1, E2F2, or E2F3) leads to the upregulation of several centrosome and/or mitotic regulators, including Polo kinase-4 (Plk4), Shugoshin-like protein 1 (Sgo1), Monopolar spindle 1 (Mps1, also known as TTK), NIMA-Related Kinase 2 (Nek2), budding uninhibited by benzimidazole-related 1 (BUBR1 or BUB1B) and serine/threonine phosphatase PP2AC.28,29 Moreover, we have found that some of these proteins are involved in EMT regulation. For example, TTK suppresses EMT and cell invasion of TNBC cell lines through the mitigation of TGF-β-induced SMAD-3 phosphorylation, induction of the transcription factor KLF5, and the dysregulation of the micro-RNAs (mi-Rs) miR-200, and miR-21. 30 Furthermore, we have found that Nek2 signals centrosome and reduces E-cadherin levels in Her2+ mammary tumors.29,31–33 Other authors have also shown a role for Sgo1 in tumorigenesis and EMT.15,34 However, no study has linked Sgo1 to EMT in TNBC.
Nek2 and TTK are kinases and can be targeted in TNBC. There is one TTK inhibitor (CFI-402257) currently in clinical trials (NCT03568422 and NCT02792465). On the other hand, there are no Nek2 inhibitors in clinical trials. However, there are some chemical inhibitors against Nek2, including INH1, which disrupts the Hec1/Nek2 interaction required for spindle assembly checkpoint function, which was shown to decrease tumor growth in athymic BALB/c-nude mice xenograft model derived from human TNBC cells MDA-MB-468. 35 Sgo1 is not a kinase, and therefore, targeting this molecule will represent a challenge. In the present review, we explore other potential targets that are mitotic kinases upstream of Sgo1 and their effects on survival and EMT in cancer with a focus on breast cancer. These kinases include Aurora A, Aurora B, Bub1, and Hec1. Many studies have reported that deregulation of these mitotic kinases causes dysregulated mitosis and aneuploidy, processes that are closely associated with genomic instability and tumorigenesis. Potential biological targets for mitotic kinases may help treat poor prognosis breast cancers, especially since inhibitors against some of these kinases (e.g. the Aurora Kinases) are already in clinical trials (NCT02719691, NCT01091428, NCT01639248, NCT03245190, NCT03216343).
Aurora A and Aurora B
Aurora kinases A and B (here forth referred to as Aurora A or B, respectively) are a family of highly conserved serine/threonine kinases that share a similar protein structure, a similar kinase activity, and are well known for their central role in regulating cell division.36,37 Aurora A is essential for bipolar mitotic spindle formation, centrosome duplication and separation, chromosomal alignment, spindle assembly checkpoint, and cytokinesis, when Aurora A is inhibited cells are arrested in G2 and their mitotic entry is blocked.36,38 Aurora B is a member of the chromosome passenger complex and regulates chromosome condensation, chromosome bi-orientation, spindle checkpoint formation, chromosome segregation, and cytokinesis.37,39 Aurora B participates in several regulatory mechanisms that maintain genome stability. For example, studies have shown that Aurora B is required in the mitotic activation of the ATM kinase, an essential kinase in DNA damage response and a mitotically activated ATM phosphorylates Bub1. 40 Studies have elucidated that ATM phosphorylation of Bub1 is required for Bub1 activity and is important for proper activation of the spindle assembly checkpoint and optimal DNA damage response.40,41 DNA damage response and spindle assembly checkpoint are critical for the maintenance of genome stability. 40 Aurora B is also involved in the inhibition of p53 transcriptional activity, which could suggest that overexpression of Aurora B may compromise its tumor-suppressive function.42,43
Aurora kinases are constitutively expressed in actively dividing cells and upregulated in highly proliferative tissues. 36 Although these kinases normally have tumor suppressor roles because of their role in the spindle assembly checkpoint, they are potential oncogenes if dysregulated, perhaps by promoting cancer cell survival and proliferation.44,45 It is well documented that overexpression or gene amplification of these kinases is associated with tumorigenesis through multiple cellular processes and has been linked to several human cancers, such as breast, ovarian, lung, and prostate. 46 A recent study based on a phosphoproteomic data analysis using the Clinical Proteome Tumor Analysis Consortium (CPTAC) in six different cancer types (breast, renal, colon, lung, ovarian, and uterine corpus endometrial) found that Aurora A and Nek2 were the most common kinase targets across these cancer types. 47 This is not surprising since Aurora A and Nek2A are structurally identical in 31%. 48 When overexpressed, Aurora A drives centrosome amplification,49–51 genomic instability and oncogenic transformation, 38 tumor cell migration, EMT transition, and metastasis. 36 EMT involves the loss of adhesion molecules and cell polarity, which confers a migratory capacity to malignant cells. Aurora A triggers EMT directly by activating the transcription activity of EMT transcription factors Twist, Slug, and ZEB. 36 Aurora A is correlated with the Wnt/β-catenin and PI3K/Akt signaling pathways; A study by Liu et al. found that MLN8237, a specific Aurora A inhibitor, suppressed the activity of the two signaling pathways and inhibited the expression of EMT-related genes, including Twist, through regulating histone modification.46,52 Meanwhile, in these MLN8237-treated cells, the expression of E-cadherin was increased and the expression of N-cadherin and β-catenin were decreased. Aurora A can also promote the expression of MMPs (e.g. MMP2, MMP7, MMP10) which are responsible for extracellular matrix degradation, facilitating the migration of cancer cells to distant sites.46,52 In nasopharyngeal carcinoma, inhibition of Aurora A by either VX-680 selective inhibitor or its downregulation with siRNA leads to suppression of CNE-2 cells invasion and restoration in the expression of E-cadherin and β-catenin epithelial markers at the cell membrane, suggesting a reversal in the EMT process. Also, suppressing Mitogen Activated Protein Kinase (MAPK) by using siRNA or MEK1/2-selective inhibitor U0126, they found that regulation of EMT by Aurora A was mediated by MAPK pathway. 53
On the other hand, overexpression of Aurora B contributes to aneuploidy and tumor progression and survival.36,46 We conducted a survival analysis and our preliminary assessment suggests that high expression of both Aurora A and Aurora B may be associated with a worse prognosis in breast cancer patients (unpublished data). Studies have elucidated the role of Aurora B in breast cancer progression by using Aurora B inhibitors leading to suppression of metastasis. For example, Zhang et al. found that elevated Aurora B expression induces the OCT4/AKT/GSK3β/Snail1 signaling since Aurora B can activate AKT by phosphorylating of OCT4 phosphorylation site (T343), which leads to inactivation of GSK3β and Snail1 stabilization to facilitate EMT and metastasis of breast cancer. 37 Their findings also showed that targeting Aurora B with an inhibitor or by its silencing inhibits OCT4/AKT/GSK3β/Snail1 signaling and reverses EMT in TNBC cells. Other studies have reported that upregulated Aurora B induced an increase in PTK2 (FAK), AKT, PI3K, and NF-ĸB protein levels which enhanced the malignant phenotype of osteosarcoma cells via activation of the PTK2/PI3K/AKT/NF-κB pathway. 54 As mentioned previously, Sgo1 is a substrate of Aurora B and its inhibition leads to genomic instability and colon tumor formation.55,56 However, it was recently demonstrated that Sgo1 is highly expressed in prostate cancer and induces proliferation and metastasis through AKT inhibition while inhibiting apoptosis through AKT. 57 It has been shown that the AKT/mTOR pathway in prostate cancer is associated with the expression of MMP9 58 and the downregulation of E-cadherin, 59 leading to EMT, invasion, and metastasis. This suggests that intermediate levels of Sgo1 are preferred to avoid tumor formation and/or progression. Given the fact that Aurora B acts as an upstream regulator of Sgo1, and that separately Aurora B and Sgo1 act through AKT raises the question of whether Aurora B and Sgo1 exert their effects in TNBC using this mechanism. We found that the role of Sgo1 in breast cancer cells is context-dependent since it can suppress CA/CIN in Her2+ cells, 60 while it suppresses CA in mammary epithelial cells overexpressing E2F1, E2F2, and E2F3. 28
The functional diversity exhibited by Aurora kinases in cancer makes them interesting targets for cancer-directed therapies, including breast cancer. Recent clinical trials are summarized in Table 1. From the trials summarized in Table 1, Aurora kinase inhibitors have gotten the farthest in clinical trials relative to other mitotic kinases reviewed here. For example, a phase II trial (identifier NCT01639248) using a dual inhibitor of Aurora A and multiple angiogenesis kinases suppressed cancer progression in 17% of metastatic TNBC patients. 61 Ongoing clinical trials targeting Aurora A are evaluating combination therapy of a highly specific Aurora A inhibitor, alisertib, with other drugs to determine how it may shrink or slow tumor growth in breast cancer patients (NCT02719691, NCT02187991). An Aurora B/Aurora C inhibitor, GSK1070916A, showed interesting results in a tumor xenograft model and a phase I clinical trial was completed but was not pursued into phase II (NCT01118611). Moreover, Chiauranib, which simultaneously targets VEGFR/Aurora B/CSF-1R, is under evaluation as a monotherapy in patients with hepatocellular and small cell lung cancer (NCT03245190, NCT03216343). Furthermore, Chiauranib has completed phase I in patients with ovarian cancer and advanced solid tumors (NCT03166891, NCT02122809).
Mitotic kinases in clinical trials.

Bub1
Bub1 is a serine/threonine kinase essential in mitotic spindle checkpoint and chromosome alignment.62,63 Bub1 functions in maintaining centrosome cohesion by phosphorylation and controlling binding sites for Sgo proteins; therefore, localizing them to the chromosome arms and centromeres, thus maintaining proper inheritance of chromosomes.64–66 Bub1 along with other conserved proteins such as Mad1-3p, Bub1p, Bub3p, and TTK ensure the fidelity of chromosome segregation.67–70 The downstream target of this complex is Cdc20, an activator of the E3 ubiquitin ligase known as Anaphase-promoting complex (also known as cyclosome or APC/C).71–73 Bub1p is required to enhance the inhibition of APC/C. 66 Despite Bub1 and Mps1 acting on Sgo1 and ensuring chromosome biorientation, they do so independently of Aurora B. 74 This suggests an important alternative pathway that signals Sgo1. Errors in chromosome segregation lead to aneuploidy, a hallmark of cancer.75,76 Bub 1 has been linked in numerous cancers to tumorigenesis by promoting cell proliferation, tumor growth, metastatic potential, and a poor patient prognosis. 77 Kaplan Meier (KM) plotter analysis from our unpublished data suggests that high expression of Bub1 may be associated with worse overall survival in breast cancer patients. Meanwhile, loss of function mutations or reduced gene amplification or expression of Bub1 has been identified in several malignancies, including colon cancer, esophageal cancer, breast cancer, and melanoma. 63 Yu et al. demonstrated that a Bub1 inhibitor may attenuate glioblastoma cellular proliferation in vitro and delayed tumor growth with prolonged survival in vivo. 77 Nyati et al. identified Bub1 as an essential mediator of TGF-β signaling, a family of cytokines that mediate many processes including immune suppression and EMT. 78 TGF-β can induce EMT when it cooperates with Ras to induce Snail or through Smad-mediated complex that indirectly regulates the expression of Snail, Slug, and Twist (reviewed in Seoane and Gomis 79 ). As previously explained, ATM phosphorylation of Bub1 is essential for the activity of Bub1 in spindle checkpoint activation and optimal DNA damage response. 40 These two cellular processes are critical in the maintenance of genome stability. 40 Bub1 nuclear localization acts as a prognostic factor in breast cancer patients. 80 The authors found that the nuclear status of Bub1 correlates with stage, pathological tumor factors, lymph node metastasis, distant metastasis, and histological grade as well as Ki67 expression. However, these associations were not found for cytoplasmic expression, suggesting that nuclear but not cytoplasmic Bub1 has an important role in proliferation and/or progression in breast carcinoma. Recently, Siemeister et al. 81 showed that inhibition of Bub1 by BAY 1816032 sensitizes tumor cells (in vitro and in vivo) toward taxanes, ATR, and PARP inhibitors. The authors reported a significant reduction in tumor size and excellent tolerability upon drug combination in tumor xenograft mice models. Despite these interesting findings, there are no current ongoing clinical trials on this drug or any other to target Bub1.
Hec1
Hec1 is a spindle checkpoint regulator that is part of the conserved NDC80 complex that regulates mitotic processes.82,83 It is responsible for recruiting checkpoint proteins to the kinetochore and ensuring proper chromosome segregation.83,84 Overexpressed Hec1 is reported in many malignancies and is associated with cancer formation, progression, and survival. 82 Accordingly, our unpublished findings suggest that overexpression of Hec1 is associated with a worse prognosis in the combined molecular subtypes survival plot for breast cancer patients. Deletions of Hec1 in tumor cell lines demonstrated its potential as a biological target by inducing mitotic abnormalities and apoptosis. 83 Inactivation of Hec1 leads to severe chromosomal errors. Limited clinical trials are available for Hec1/NDC80 in cancer. A phase II clinical trial is assessing the proliferation gene signature based on 11 highly correlated signature genes, including NDC80, in TNBC patients. Moreover, Hec1 interacts with TTK through a mechanism in which Aurora B phosphorylates the N-tail of Hec1 to destabilize the kinetochore-microtubule attachment, which ultimately enhances the localization of TTK to the kinetochore, demonstrating the direct binding of TTK kinetochore localization domain with the Hec1 microtubule-binding domain. 85 Hec1, which is also a serine phosphoprotein, binds to Nek2 during the G2/M phase. 86 Nek2 is a serine/threonine that localizes to the centrosome, phosphorylates Hec1 to control kinetochore attachment, alignment, and signaling of the spindle assembly through Mad1 and Sgo1.15,87 Nek2 has a myriad of roles in the cell cycle and tumor progression beyond Hec1 phosphorylation. Nek2 is highly overexpressed in several cancer types (including breast cancer) and correlates with tumorigenesis, tumor progression, and chemotherapy resistance. 15 Moreover, Nek2 has been shown to correlate with EMT in hepatocellular cells modulating E-cadherin, N-cadherin, and Vimentin 88 as well as MMP9. 89 Several other studies have shown that Nek2 induces EMT through β-catenin.90,91 Despite the myriad roles of Nek2 in cancer, currently, there is no inhibitor in clinical trials for Nek2. Yet, phosphorylation of Hec1 by Nek2 kinase is essential for its mitotic function; therefore, a disruption in Hec1/Nek2 perhaps will have potential for cancer therapy. 82 Likewise, Sgo1 is phosphorylated by Nek2 for essential faithful chromosome segregation and the proper attachment of the spindle microtubule to the kinetochore. 92 This denotes the redundancy in the cell, which can be advantageous to target several kinases to improve and diversify treatment options for TNBC patients.
Conclusions
As reviewed in this work, along with their roles as important proteins controlling cell cycle activity, mitotic kinases are involved in breast cancer progression by altering processes including proliferation, EMT, and metastasis, thus potentiating their role in breast cancer aggressiveness. This work reviewed the clinical trials of Aurora A, Aurora B, Bub1, and Hec1, from which Aurora A showed to induce EMT by controlling EMT and MMPs biomarkers. On the other hand, inhibition of Aurora A leads to the suppression of EMT through the downregulation of the MAPK pathway. Targeting Aurora B inhibits the EMT and suppresses cancer progression, while both Aurora B and Nek2 regulate Hec1 for the maintenance of mitotic function. Bub1 controls TGF-β which induces EMT. All these mitotic kinases induce EMT directly or indirectly, thus contributing to cancer progression (Figure 1). Therefore, the need for more research and the development of inhibitors to target these mitotic kinases are highly important to the incorporation of novel treatments for TNBC.
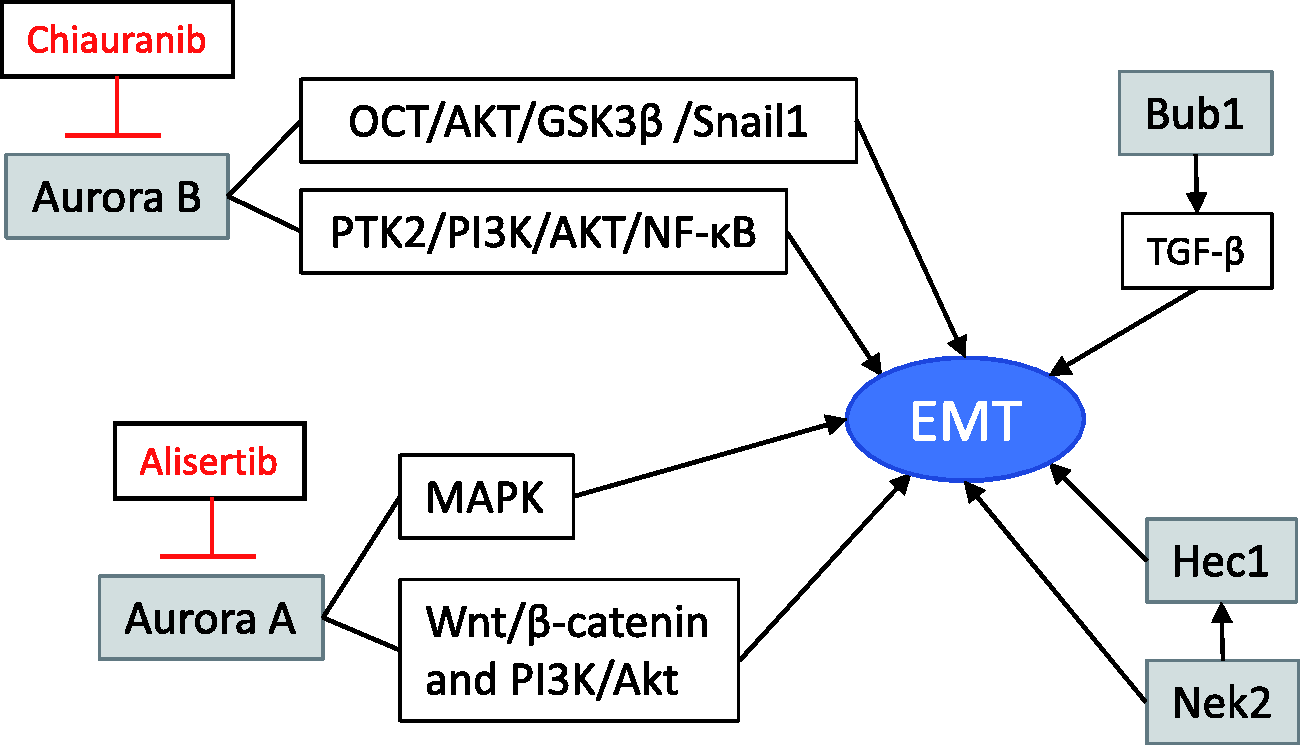
Model portraying how mitotic kinases connect to control epithelial-to-mesenchymal transition (EMT), invasion, and metastasis in breast cancer. Dysregulation of mitotic kinases activates a series of events that ultimately lead to EMT and metastasis allowing them to become potential oncogenes. Aurora A directly triggers EMT by activating the transcription factors Twist and Slug. Aurora A correlates with Wnt/β-catenin and PI3K/Akt signaling and was found to lead to EMT progression. Other studies have found that Aurora A regulated EMT is mediated by MAPK phosphorylation. Similarly, inhibition of Aurora A leads to the suppression of EMT through the downregulation of the MAPK pathway. Targeting Aurora B inhibits the OCT/AKT/GSK3β/Snail1 signaling, reverses EMT, and suppresses cancer progression, while both Aurora B and Nek2 regulate Hec1 for the maintenance of mitotic function and cell survival. Phosphorylation of Hec1 by Nek2 is essential for its mitotic function. Nek2 may signal EMT through Hec1 or independently of Hec1. Bub1 controls TGF-β which induces EMT. Inhibitors in clinical trials are shown in red. (A color version of this figure is available in the online journal.)
Footnotes
Authors’ contributions
The general concept and editing were done by HIS. The literature search, specific sections, concepts, writing, and design of the figures were done by SC-M. SJ and YR-R contributed to the literature search, writing, and editing of the manuscript.
DECLARATION OF CONFLICTING INTERESTS
The author(s) declared no potential conflicts of interest concerning the research, authorship, and/or publication of this article.
FUNDING
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the PHSU-MCC Partnership grant #2U54-CA163071 from the National Institute of Health, the Puerto Rico Science and Technology Trust Advanced grant and the Hispanic Alliance for Clinical and Translational Research (Alliance) #U54GM133807. The project was also supported by the National Cancer Institute F31 NRSA Fellowship Award #1F31CA247459 corresponding to SJ.