
Abstract
Cerebral palsy (CP) is a lifelong disability characterized by the impairment of brain functions that result in improper posture and abnormal motor patterns. Understanding this brain abnormality and the role of genetic, epigenetic, and non-genetic factors such as signaling pathway dysregulation and cytokine dysregulation in the pathogenesis of CP is a complex process. Hypoxic–ischemic injury and prematurity are two well-known contributors of CP. Like in the case of other neurodevelopmental disorders such as intellectual disability and autism, the genomic constituents in CP are highly complex. The neuroinflammation that is triggered by maternal cytokine response plays a critical role in the pathogenesis of fetal inflammation response, which is one of the contributing factors of CP, and it continues even after the birth of children suffering from CP. Canonical Wnt signaling pathway is important for the development of mammalian fetal brain and it regulates distinct processes including neurogenesis. The glycogen synthase kinase-3 (GSK-3) antagonistic activity in the Wnt signaling pathway plays a crucial role in neurogenesis and neural development. In this review, we investigated several genetic and non-genetic pathways that are involved in the pathogenesis of CP and their regulation, impairment, and implications for causing CP during embryonic growth and developmental period. Investigating the role of these pathways help to develop novel therapeutic interventions and biomarkers for early diagnosis and treatment. This review also helps us to comprehend the mechanical approach of various signaling pathways, as well as their consequences and relevance in the understanding of CP.
Impact Statement
Cerebral palsy (CP) is a permanent disability that is defined by brain function impairment that results in improper posture and aberrant movement patterns. Prematurity and hypoxic–ischemic damage are two well-known factors contributing to the development of CP. It has been discovered that the neuroinflammation caused by maternal cytokine response plays an important part in the pathogenesis of fetal inflammation response, which is one of the contributing causes for CP, and it persists even after the delivery of children with CP. The canonical Wnt signaling pathway is crucial for mammalian fetal brain development and it governs many processes such as neurogenesis. The antagonistic activity of GSK-3 in the Wnt signaling pathway plays an important role in neurogenesis, neural development, and brain development.
Introduction
Cerebral palsy (CP) is a non-progressive neurodevelopmental disorder that affects approximately one in five hundred children. Its clinical features include impairment in movement and posture, motor deficiency, lack of coordination, and upper motor neuron disorder involving spasticity. 1 The motor impairment of CP is often accompanied by other related impairments such as epilepsy, intellectual disability, and sensory disorders. Although numerous risk factors associated with CP are identified, for many individuals, it is difficult to determine the etiology of CP since it involves multiple mechanisms. Some studies report that acute intrapartum hypoxic-ischemia, which accounts for less than 10% of cases, is the main cause of CP.2,3 The risk factors associated with CP include congenital malformations, multiple gestations, preterm birth, intrauterine inflammation and infection, birth asphyxia, thrombophilia, and perinatal stroke. An important pathophysiological mechanism that is observed is the infection of amniotic fluid and intra-amniotic inflammation that causes damage to the developing brain of the fetus, leading to CP. This damage may persist for many years. Several neurological and neuropsychiatric disorders are associated with perinatal infection and inflammation, and these are found to have long-term consequences on the child’s brain. 4 Brain encephalopathy in term-born infants accounts for 24% of all cases and is reported to cause inflammatory responses in the first week after birth, which is correlated with the level of brain injury.5,6 Preterm deliveries affect approximately 15 million births 7 and are found to be important risk factors for many congenital disorders including cognitive impairment, CP, autism, and mental disorders that are discovered later in life. 8
Several studies investigated that magnetic resonance imaging (MRI) of cerebral phenotypes of preterm babies are associated with later functions and involves dysregulation of neural pathways, structural alterations of cerebral cortex and gray matter, and diffused white matter diseases.9–12 Many studies indicated that there has been a decline in the incidence rate of CP over the last few decades due to improved prenatal care and obstetric practice. These results led some researchers to investigate the “unknown pathophysiologic approaches” that account for significant proportion of CP. 13 Out of the studies conducted, some of them suggested that the unknown pathophysiology of this disease may include genetic factors and dysregulation of some neuronal pathways. In this article, we investigated the association of genetic or epigenetic factors, neuronal signaling pathways, and inflammatory response alterations with CP. Understanding these pathways can aid in the development of novel, effective, and safe therapeutic treatments for CP.
Methodology
This study aimed to investigate the clear insights about the mechanistic pathways involved in the causation of CP as well as the food pattern and nutritional status of children affected with CP. First, we have identified the research articles with appropriate words and phrases. We have used search engines like PubMed, Google Scholar, and MEDLINE. The key phrases used were “cerebral palsy” and “signaling pathways involved in cerebral palsy” combined with “nutritional status” and “food intake in CP children.” Total number of papers identified (n = 500). The inclusion criteria for paper selection were that we have identified papers including studies related to multiple signaling pathways and nutritional status of children with CP. Total number of papers after inclusion–exclusion consideration (n = 150) and full-text screened researched articles (n = 100). Total number of papers included into the study (n = 75).
Genetic or epigenetic implications in CP
Current research investigates that 30% of CP cases are caused by genetic or epigenetic factors.14–16 Four main types of DNA variations are identified that contribute to the pathogenesis of CP. The unfortunate outcome of the many mutations is that it results in the loss of important cellular functions of several proteins that are encoded by these genes. Figure 1 depicts the contribution of genetics in the pathophysiology of CP.
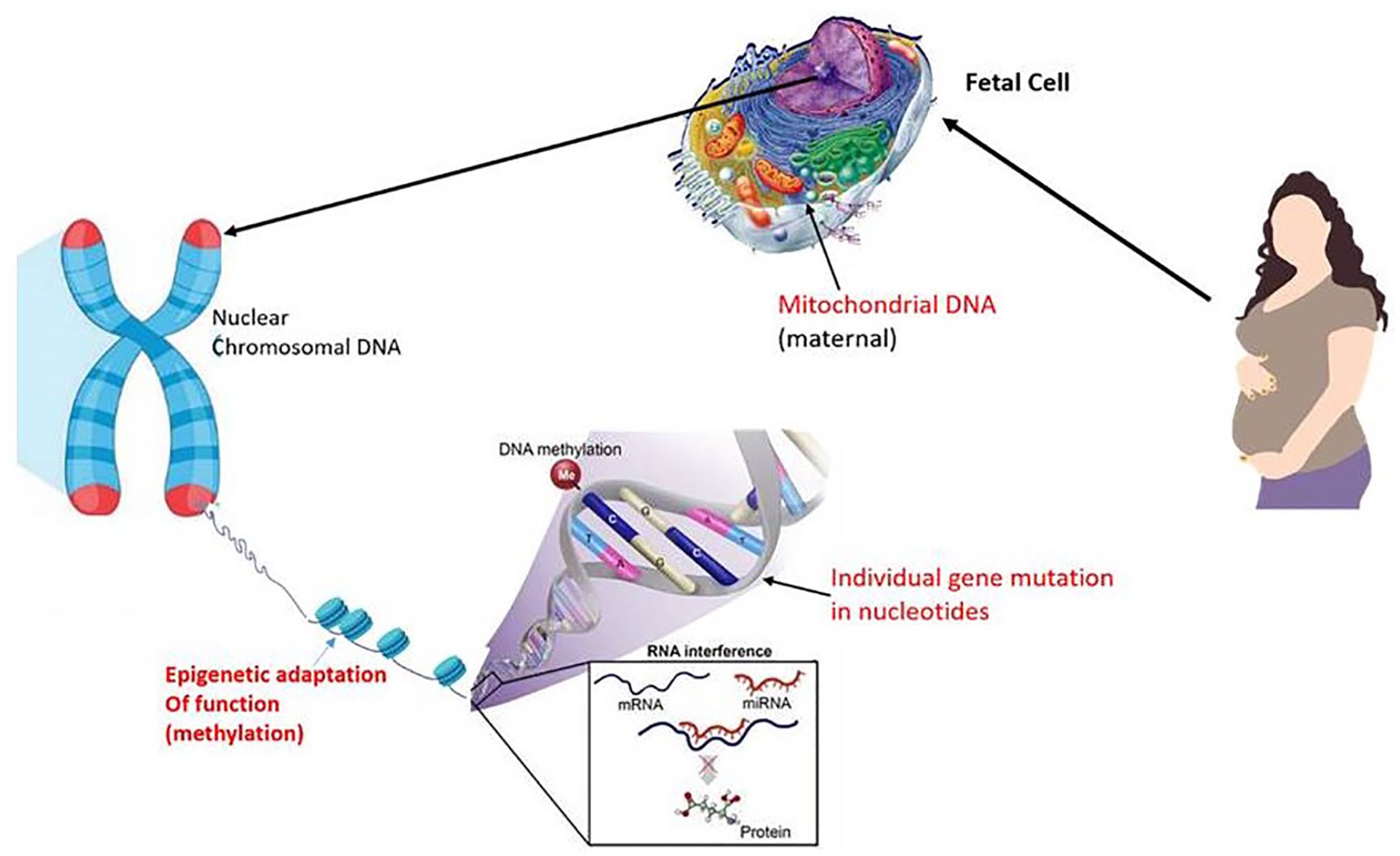
Genetic implications in cerebral palsy showing main sites and types. (A color version of this figure is available in the online journal.)
The effects of genetic mutation may vary depending on its nature, involvement of environmental factors, and the individual genomic profiles where mutation occurs. A mutation can either be severe, where the deleterious mutation causes major effects, or minor, where the damage caused is less enough to not affect the protein function. A deleterious mutation is sufficient to cause CP in most individuals, while in others cases, minor mutations coupled with environmental factors like hypoxia may cross thresholds and cause severe neuromotor disorders. For instance, CP is not caused by one single mutation, but the combined effect of several less damaging gene mutations in a polygenic manner. 17 A study conducted in hypoxic–ischemic rabbit model that is induced by aortic ligation revealed that when their litters were exposed to the same insult, they developed motor impairment suggesting that genetic or epigenetic modulators may mediate adverse motor outcome and extent of injury. 18 Mitochondrial DNA is suggested to be a potential biomarker for many dysfunctions of mitochondria, and it is found to be related to many diseases including neurological diseases, traumatic brain injury, and autism. Lu et al. 19 investigated the association of mitochondrial DNA copy number with CP and reported that a decline in mitochondrial DNA copy number implied mitochondrial dysfunction and CP. A study that was conducted reported that the mutation in mitochondrial DNA 8993 is associated with Leigh encephalopathy in children, where CP or other neurological impairment was found in other family members. These family members were examined to analyze the correlation between mutant mitochondrial DNA and the severity of the disease. 20 It was observed in 10–20% of the cases that genomic copy number variations, which involve deletions or duplications, may cause CP.21–23 Microarray techniques were used by researchers on a cohort of 52 subjects suffering from cryptogenic CP. In this study, the ACMG (American College of Medical Genetics and Genomics) standard was applied to interpret the results where it was identified that 31% of cases were pathogenic or likely to be pathogenic copy number variation. 22 Another study used the same technique on 147 non-selected CP patients and found copy number changes that met ACMG requirements. 23 Epigenomic studies revealed some alterations in axon structure, actin cytoskeleton, and cell signaling, which was aligned with genomic studies. Alterations caused by DNA methylation that show changes in mitogen-activated protein kinase (MAPK) signaling, inflammation, cytokine–cytokine receptors, and Ras signaling at birth were observed in monozygotic twins discordant for CP. 24 In non-selected subjects with CP, there were differences identified in the methylation process in the actin cytoskeleton, axon structure, crosstalk between dendritic cells and natural killer cells, insulin receptors, neuregulin, transforming growth factor beta (TGF-β), Wnt signaling, and phosphoinositide 3-kinases (PI3Ks)/Akt signaling pathways. 25 Many cellular signaling pathways that involve synaptic dysfunction, activity-dependent translation and transcription, neuroglia signaling, and neuroinflammation are dysregulated in numerous neurodevelopmental disorders, which necessitate in-depth research by utilizing animal models. It was seen that transcriptomic studies of the lymphoblastoid cell lines of CP subjects (n = 182) who are exposed to genetic, environmental, and indeterminate causes reported 387 differentially expressed genes. 26 An analysis of these gene pathways demonstrates a down-regulation of cell signaling and transduction including an upregulation of immune system-related genes, brain-derived neurotrophic factor (bdnf), and altered amyloid precursor protein A. Some studies suggested an overlap in dysregulation of MAPK signaling and result of the epigenetic analysis. A report showed that defects in genes that regulate cell-signaling pathway – that is, MAPK, PI3K, or Akt – may cause CP and neuronal signaling defects. 26
Implications of cytokine dysregulations in CP
While one of the etiologies behind the CP is neuroinflammation that is triggered by several mechanisms including maternal/fetal infection, hypoxia, maternal preeclampsia, and stroke, the other possibility is genetic etiologies. 27 The cytokine response that is observed in a maternal amniotic fluid includes increased levels of interleukins (IL-6 and IL-1β) and tumor necrosis factor (TNF) alpha. In the fetal compartment, the level of IL-6 is found to play an important role in the pathogenesis of inflammatory syndrome in developing fetuses, which is associated with CP. This inflammatory pathway appears to continue even after the birth of CP subjects and gives rise to the “sustained inflammation hypothesis” that suggests that prenatal, antenatal, or neonatal pro-inflammatory cytokines induce inflammation that contributes to dysregulation of cytokine pathways. 28 A study conducted on school children having post-neonatal encephalopathy (NE) explored the cytokine response to report an abnormally elevated level of cytokines in these children. The level of granulocyte-monocyte-cerebrospinal fluid (GM-CSF), IL-6, IL-8, and TNF-β was found to be significantly high in children with NE when compared to control subjects. IL-8 and GM-CSF were found to be significantly elevated in children with NE upon stimulation with LPS (lipopolysaccharides) when compared with age-matched controls. Hypo-responsiveness of LPS in various cytokines among schoolchildren demonstrates an altered immune response. 29 A study conducted by Huang et al. 30 investigated the association of umbilical cord blood cytokines with CP in premature babies. They performed enzyme-linked immunoassay technique and identified a significantly high level of IL-8, PGE2, and myeloperoxidase (MPO) level in preterm babies with a gestation period of 32 weeks when compared to full-term babies. These cytokines are not related to gestational age but to preterm birth. It was also seen that cytokine IL-8 was increased in CP-affected preterm infants but not MPO. Some of the evidence suggested that preterm deliveries caused by cytokine induction are mostly due to chorioamnionitis. It was observed that intrauterine inflammation or infection that caused activation of cytokines and elevated the level of pro-inflammatory cytokines in amniotic fluid and neonatal blood that the preterm baby was exposed to was identified as an important reason for preterm deliveries, CP, and periventricular leukomalacia (Figure 2). 31 One study evaluated the association of four TNF-α promoter single nucleotide polymorphism (SNPs), two IL1β SNPs, and one IL-6 polymorphism that is susceptible to CP in preterm babies. Their results investigated IL-1β and TNF-α polymorphism that is related to a higher level of cytokines in the circulation, to find their role in genetic susceptibility to damage white matter and cause CP in preterm infants. 32 Animal model studies provided evidence that ischemic injury and inflammation/infection-induced brain injury play a major role in CP pathogenesis. A study that was based on reverse transcriptase polymerase chain reaction (PCR) methods showed elevated levels of pro-inflammatory cytokines including IL-6, IL-1β, and MCP-1 (monocyte chemoattractant protein-1) in the brains of mouse pups that were exposed to in-utero lipopolysaccharides (LPS). 33 Some studies revealed that a dose-dependent elevation was observed in the expression of TNF-α and IL-1β mRNA in rat fetal brain that are exposed to in-utero LPS. In addition to this, the hippocampus and cortical region of the brain observed a significant decrease in the level of myelin basic protein, elevations in the level of glial protein (acidic or fibrillar), positive astrocytes, and changes in immune reactivity of oligodendrocytes (OLs). 34 Table 1 represents the association of inflammatory cytokines with CP.35–48
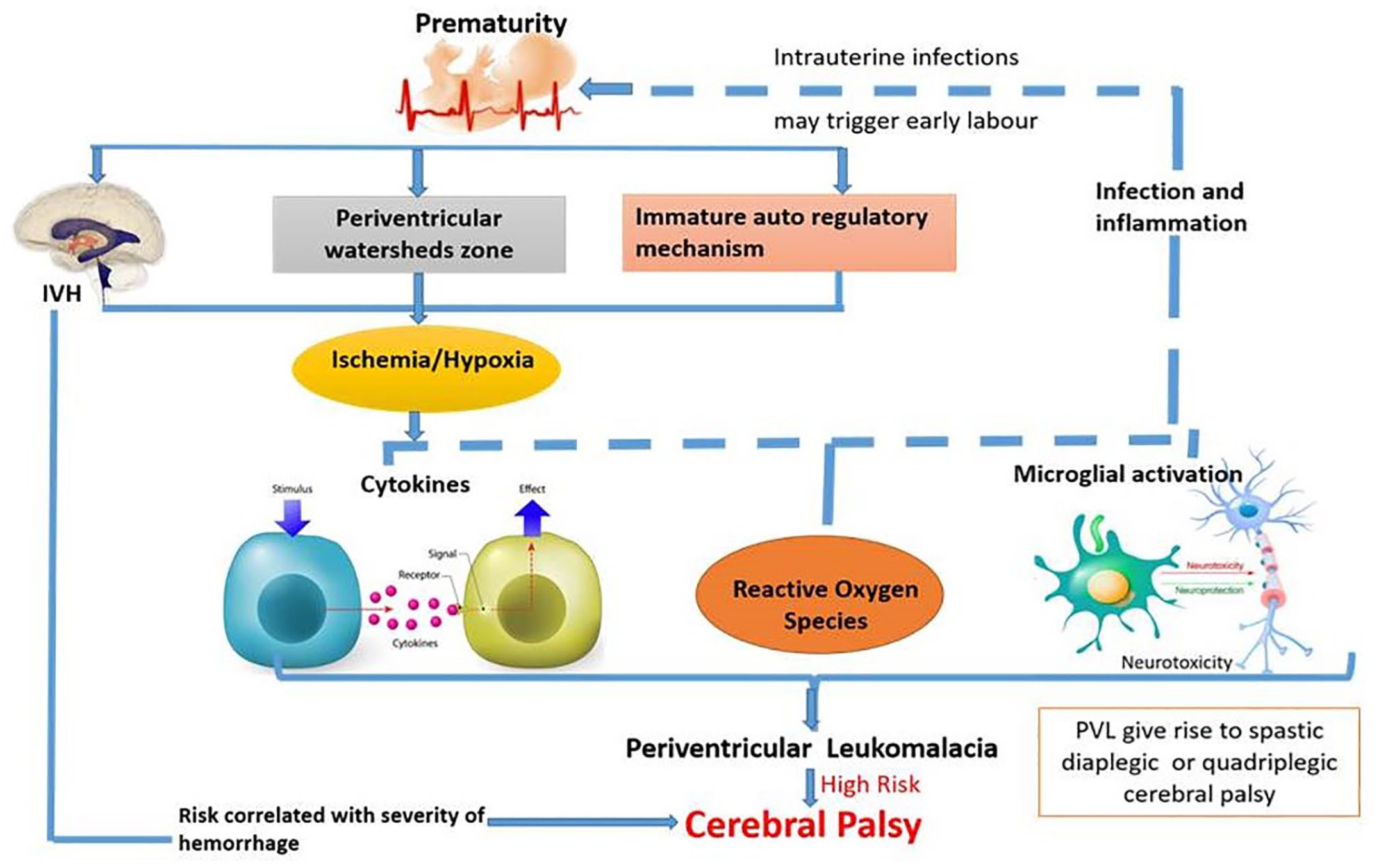
Association of cytokines, reactive oxygen species, and maternal infection and inflammation that causes microglial activation to the prematurity and periventricular leukomalacia that are responsible for cerebral palsy. (A color version of this figure is available in the online journal.)
Association of inflammatory cytokines with CP.
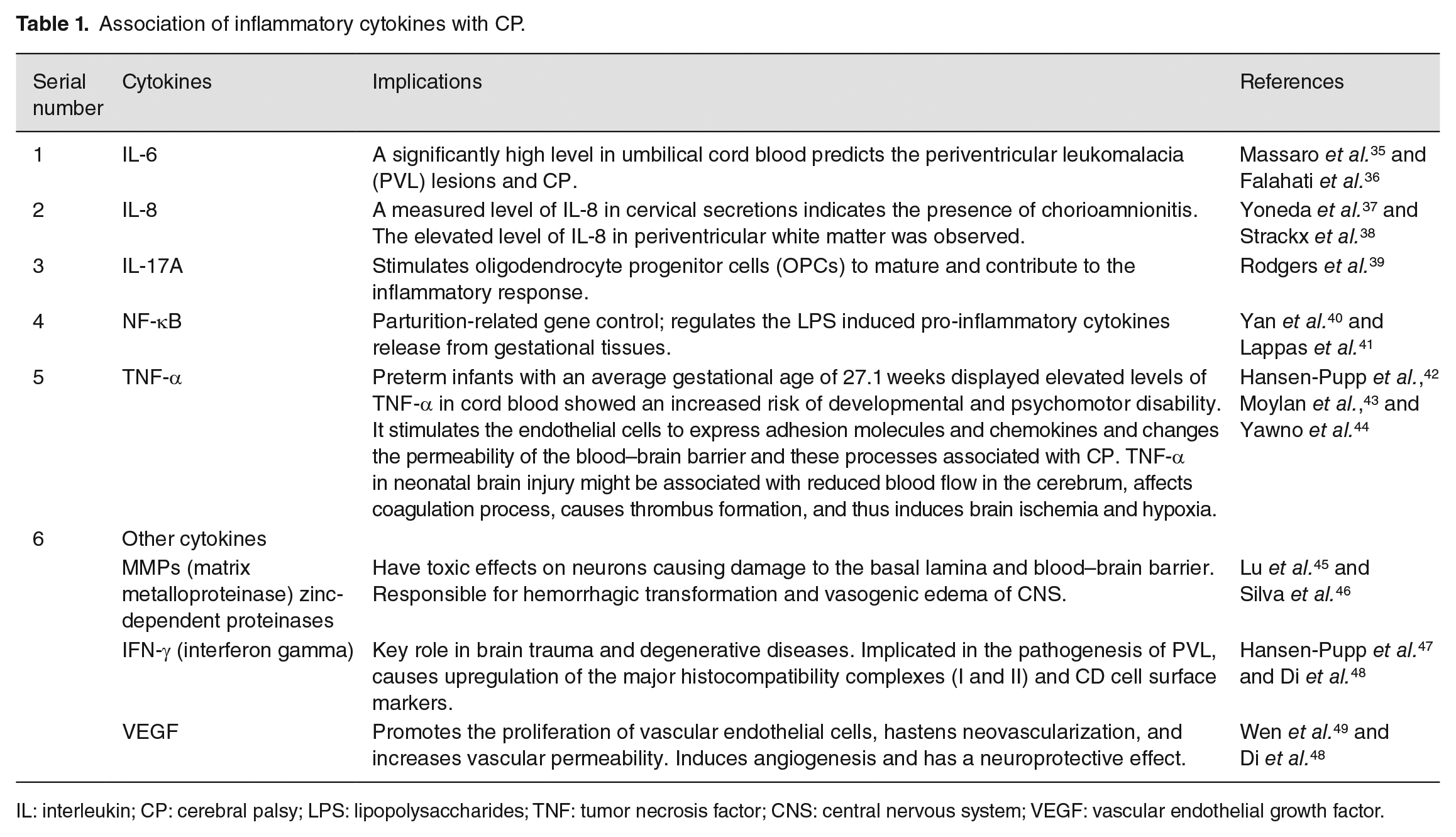
IL: interleukin; CP: cerebral palsy; LPS: lipopolysaccharides; TNF: tumor necrosis factor; CNS: central nervous system; VEGF: vascular endothelial growth factor.
Some studies reported that immune abnormalities have a strong relationship with CP and erythropoietin (EPO) plays a neuroprotective role in cell injuries associated with CP. EPO is a glycoprotein containing 165 amino acids, and is known as pleiotropic cytokine. Some studies suggested that EPO performs some non-hematopoietic actions like protection, maintenance, and development of nervous system. EPO in brain is normally secreted from astrocytes and EPO receptors are expressed principally in neurons. EPO acts as neuroprotective as well as anti-inflammatory agent by activating janus kinase 2 (JAK2)/signal transducers and activators of transcription 5 (STAT5) pathway. EPO binds with erythropoietin receptor (EPOR) in the extracellular domain causes conformational changes of receptor homodimers and results in rapid phosphorylation of tyrosine residues of JAK2 and its activation in turn recognized by several Src homology-2 (SH-2) domain containing signal transduction molecules involving STAT5. Activation of this pathway in turn activates p85 subunit of PI3PK, NF-β, MAPK, and STAT5. STAT5 phosphorylation causes translocation and dimerization of the nucleus by acting as transcription factor and regulates the expression of various EPO-responsive genes. STAT5 is dephosphorylated by intra nuclear tyrosine phosphatase, and thus terminates the process of signal transduction. Thus, EPO provides neuroprotection by activating JAK/STAT pathway. Hypoxia inducible factor-1 (HIF-1) activated during inflammation enhances the secretion of EPO. It plays the role of anti-inflammatory agent by (1) inhibiting the expression of IL-1 and IL-6 induced by ischemia; (2) stimulating inflammatory cell death by pathways including phosphatidylserine exposure, protein kinase B, and activated microglia; (3) reducing oxidative stress, inflammatory response and myelin basic protein during immune reaction. 49 The cytokine profile study in postnatal childhood revealed key mediators of cell injury in CP and provided a better understanding of its pathophysiology that help to develop novel therapeutic interventions.
Implications of PI3K–Akt–Wnt pathway in CP
The Wnt signaling pathway is important for cell patterning, regulation of stem cells, and cell cycle during the mammalian fetal growth and development including the brain.50,51 Among adults, the Wnt pathways affect the regeneration and regulation of many tissues by homeostasis and proliferation of stem cells. 52 It also maintains critical processes like axon remodeling, neuritic outgrowth, neurogenesis, and synaptic plasticity. Canonical Wnt signaling pathway suppresses glycogen synthase kinase-3 (GSK-3) and stimulates downstream regulation of signaling. While inhibiting the functions of canonical pathway of Wnt,53,54 GSK-3 also regulate other pathways that are involved in the development and function of neurons. Figure 3 represents the GSK-3 regulation of the Wnt and Akt signaling pathways, neurotrophic growth factor that activates Akt signaling to phosphorylate GSK-3 and suppresses it by allowing downstream effectors activation to promote cell survival. Neurogenesis is promoted by Wnt genes that are activated by β-catenin stabilization in Axin complex that is caused by the inhibition of GSK-3 by Wnts. It is observed that lithium antagonizes GSK-3 pools in both Wnt and Akt signaling and activate the pathway to function. Therefore, lithium activates the process of neurogenesis through Wnt/β-catenin signaling activation, and thus enhances cell survival. In the absence of Wnt ligands, GSK-3, APC (adenomatous polyposis coli), which is a key tumor suppressor gene, and β-catenin, which is a transcriptional co-activator, bind directly to the Axin protein complex to facilitate the β-catenin phosphorylation by GSK-3 that causes proteasome-dependent degradation by targeting β-catenin. Binding Wnt ligands to frizzled receptors activates the phosphorylation of LRP5 (low-density lipoprotein receptor-related protein-5 and 6) and co-receptors that cause inhibition of GSK-3 and stabilization of β-catenin. The stabilized form of β-catenin enters the nucleus and interacts with transcription factors like LEF (lymphocyte enhancer factor or T-cell factor [TCF]) family to stimulate transcription. 55
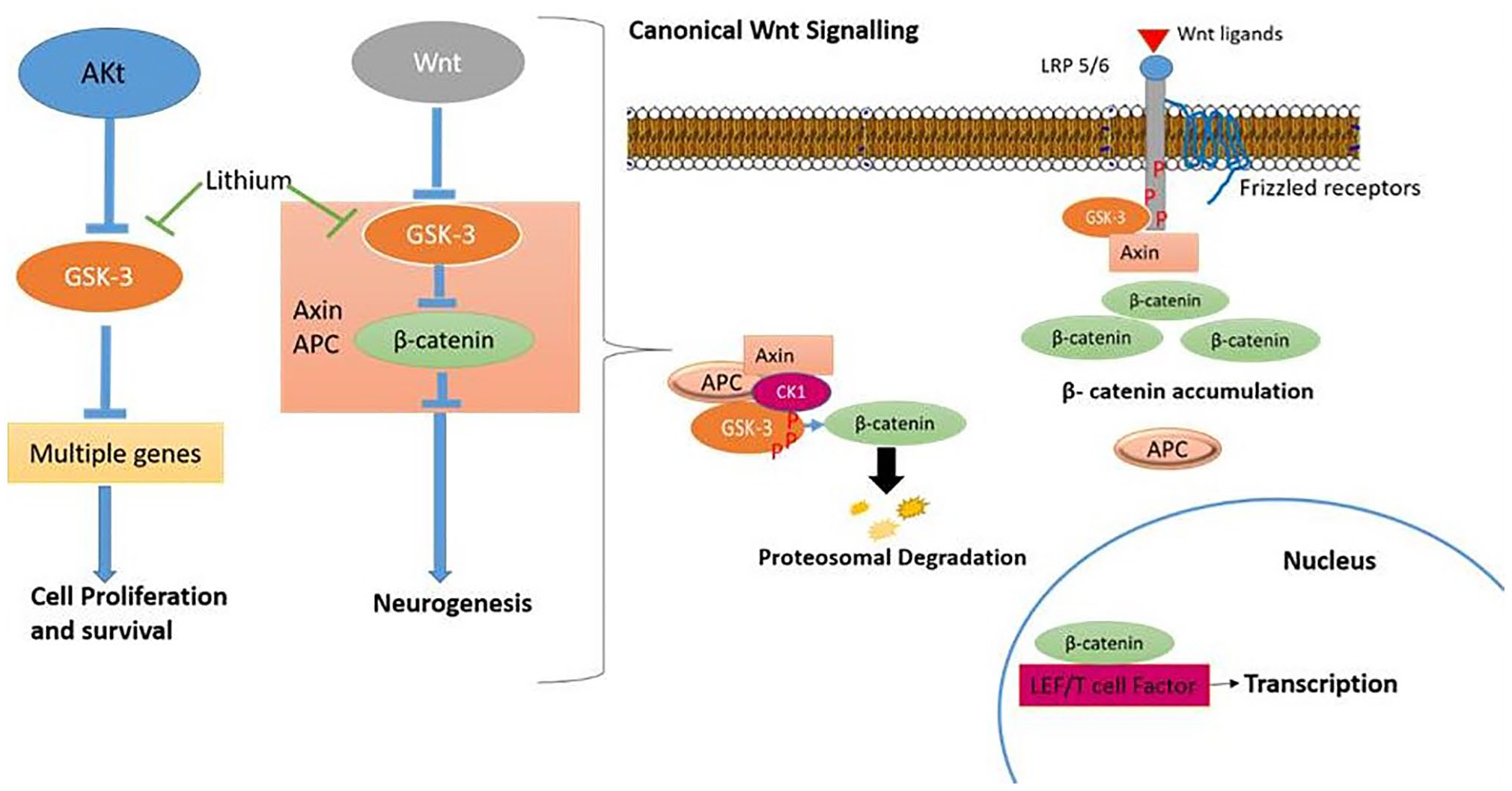
GSK-3 regulations of Wnt and Akt signaling pathways. (A color version of this figure is available in the online journal.)
Not only does GSK-3 regulate the Wnt signaling pathway, but it also regulates other signaling pathways including growth factor, notch, and sonic hedgehog pathway through Akt, thereby affecting the cell survival in the brain. 51 Neutrophils and growth factors, along with insulin, stimulate Akt and PI3K (phosphatidylinositol-3-kinase). This causes phosphorylation of GSK-3 at N-terminal of serine residues (GSK-3β at Ser9 and GSK-3α at Ser21) to form pseudosubstrate motif that inhibits GSK-3 and allows downstream effector activation such as mTOR (mammalian target of rapamycin) and glycogen synthase. 56 Significantly, it was suggested that GSK-3 in Axin complex of Wnt pathway was not regulated by the phosphorylation of serine residues at N-terminal, Wnt ligands, which were neither neurotrophin nor insulin/Akt, that induced phosphorylation of Ser9 or 21 of GSK-3 was associated with Axin complex. 57 In addition to this, the GSK-3 pool associated with Akt and Wnt signaling responses was regulated by distinct mechanisms and subcellular pools that were demonstrated in double knock-in mice, where the phosphorylation of GSK-3bser9 and GSK-3aser21 sites was mutated to alanine. 58
During embryonic development, Wnt signaling plays a critical role in the development of neural tube. It suppresses the anterior and promotes the posterior development of the neural tubes. Thus, while the inhibition of Wnt signaling pathway causes a reduced posterior development and enhanced anterior development, aberrant Wnt pathway stimulation causes reduced anterior development and enhanced posterior. 59 In continuation with this process, Wnt antagonists such as DKK1 cause anterior localization and is important for the anterior development of neural tube.59,60 In the later stages of development, Wnt signaling causes patterning of neural tube by generating signal centers in the hindbrain including creating a midbrain–hindbrain boundary and restricting rhombomere boundaries.59–61 Wnt signaling is also important for neural tube patterning in dorsal or ventral form. Wnt1 and Wnt3a expressions occur in the dorsal neural tube, and if they are deleted, the ventral cell will expand at the cost of dorsal fates. 62 Overexpression of genes Wnt1 or Wnt3a causes extension of dorsal fates. 62 Wnt signaling is important for neural crest specification since it promotes cell fate in dorsal and suppresses ventral cell fates in telencephalon during embryonic development. 59
The studies showed that Wnt signaling was important for the proliferation of precursor cells of a neuron during brain development. It was observed in a study that during the development of chick neural tube, overexpression of Wnt1 and Wnt3a, along with β-catenin stabilization, caused an increase in proliferation of neural precursor cells, whereas negative expression of dominant TCF4 resulted in decreased cell proliferation. 63 Another study in mice indicated that overexpression of Wnt1 induced neuronal cell proliferation and size expansion in the caudal midbrain area that resulted in substantial midbrain enlargement. 64 In addition, the loss of β-catenin function in the mesencephalon, diencephalon, and hindbrain caused a decrease in the size of the midbrain by reducing the progenitor cell domain, while gain of β-catenin function caused an increase in brain size by expanding the progenitor cell domain. 65 The loss of Wnt3 function caused a reduced proliferation of hippocampal neural progenitor cells and interrupted the development of the hippocampus region. 66 A similar type of defect was observed in the dorsal telencephalon when β-catenin was deleted. 67 All these studies suggested that the Wnt/β-catenin signaling process stimulated progenitor cellular proliferation for the development of neural tube, midbrain, and hippocampus during the fetal growth development period.
The research deduced that apart from patterning and cellular proliferation, Wnt signaling promoted neurite development and caused an impact on axonal size, branching, remodeling, and complexity. 68 It was observed that Wnt7a expression in cultured granular cells of the cerebellum caused an increase in axon size, neurite growth, and growth cone size while earlier studies observed that Wnt antagonist secreting frizzled receptor associated protein caused axonal remodeling and reduced growth cone size.69,70 Some of the studies also suggested that GSK-3 inhibitors were involved in promoting neurite size, outgrowth, and formation of an axon, axon size, and branching in several different cell types including granular cells of the cerebellum, neurons of dorsal root ganglion, and hippocampus.69–71 It was also observed that the Wnt in spinal cord development maintained the direction of commissural axon after it passed through the midline. This activity relied upon the aPKC (atypical protein kinase) and PI3K, but not on LTP6, which indicated non-involvement of canonical Wnt/β-catenin pathway. 72 Moreover, it was deduced from the study that Wnt and β-catenin facilitated the growth of dendrites and their branching, synaptic formation, and plasticity in the cerebellum.53,73 These studies suggested the critical role of the Wnt signaling pathway in mammalian brain development including neurogenesis. Hence, it was deduced that any dysregulation of these pathways could cause several neuro-impairment disorders like CP, autism, and others. Therefore, for developing any novel therapeutic interventions for the treatment of these disorders, further research was required in this area.
Nutritional status and food pattern of children with CP
Association of CP with comorbidities like malnutrition, gastrointestinal (GI) symptoms, impaired growth and development, epilepsy, intellectual disability was observed in many case studies.74,75 Motor dysfunction in CP causes oropharyngeal dysphagia. This may reduce food intake and consequently causes malnutrition, lung infection and pulmonary aspiration. 76 One study reported that many children with CP considered “taste” to be very important in their meals, other hemiplegic group suggested “nutrition” to be the most important. Paraplegic children and children with severe brain injury usually preferred “sweet taste” while quadriplegic children preferred greasy taste. Children with CP engaged in very few outdoor activities; therefore, they have poor synthesis of vitamin D, and intake of calcium and vitamin D is important. 77 Decreased food intake along with anticonvulsant medications causes decline in the level of bone mineral density. This results in functional impairment, muscular weaknesses, and pathological bone fractures. Inadequate food intake causes macro- and micro-nutrient deficiencies, especially anemia, vitamin A, and vitamin B complex deficiencies. The “Quality of Life” of the children with CP and their caregivers were suboptimum; correction of nutritional deficiencies especially vitamins A, D and B complex, anemia, and mental and physical support is suggested for the well-being of children suffering from CP. 78
Future perspectives and conclusion
Genetic etiologies are found to be an important contributor to the development of CP, particularly through impaired brain development and dysregulation of several signaling pathways in response to associated risk factors. The number of recurrent genes provides us strong evidence in understanding the pathophysiology of CP. Limited studies are available that determine the genetic etiologies related to motor type CP because of phenotypic and genotypic heterogeneity. Limited availability of animal model functional studies impedes the understanding of CP pathophysiology. Understanding the association of pro-inflammatory cytokines such as IL-6, IL-1β, and TNF-6 will help in developing important predictive biomarkers for detecting neurodevelopmental disorders. Regulation of Wnt signaling, β-catenin, and GSK-3 pathways suggests that the Wnt/β-catenin signaling process stimulates progenitor cellular proliferation in the development of neural tube, midbrain, and hippocampus during the fetal growth development period. Any impairment of these pathways leads to several neurodevelopmental diseases including CP and autism. A better understanding of all these mechanisms will enable us to identify the risk factors that contribute to CP. Together with the discovery of genetic factors, such as epigenetic and copy number variants, and cytokines regulation, Wnt signaling pathways can provide new opportunities for further detailed analysis and study-driven interventions to improve the lives of children living with CP.
Footnotes
Authors’ Contributions
J.U., A.S. and M.N.A. had the idea for this study and designed it and took responsibility for the integrity of the data. J.U. and M.N.A. contributed in the writing of the report. A.S. and M.N.A. contributed to the critical revision of the manuscript. All authors reviewed and approved the final version of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.