
Abstract
SINE-VNTR-Alus (SVAs) are the youngest retrotransposon family in the human genome. Their ongoing mobilization has generated genetic variation within the human population. At least 24 insertions to date, detailed in this review, have been associated with disease. The predominant mechanisms through which this occurs are alterations to normal splicing patterns, exonic insertions causing loss-of-function mutations, and large genomic deletions. Dissecting the functional impact of these SVAs and the mechanism through which they cause disease provides insight into the consequences of their presence in the genome and how these elements could influence phenotypes. Many of these disease-associated SVAs have been difficult to characterize and would not have been identified through routine analyses. However, the number identified has increased in recent years as DNA and RNA sequencing data became more widely available. Therefore, as the search for complex structural variation in disease continues, it is likely to yield further disease-causing SVA insertions.
Impact Statement
This review provides a comprehensive overview of disease-associated SVAs identified to date discussing the different mechanisms through which these insertions act. These complex structural variants can be difficult to detect and characterize through routine analysis; however, the number of disease-causing SVAs is increasing. This review highlights the importance of evaluating this type of variation when looking to identify disease-causing variants, particularly in cases where standard pipelines have been unable to determine the causative variant. Understanding the genetic component of disease could potentially lead to novel therapeutic targets.
Introduction
Though previously considered “junk DNA,” mobile DNA, or transposable elements (TEs), are a source of both genetic variation1–4 and gene expression modulation.5–7 Class I TEs, or retrotransposable elements (RTEs), utilize a copy and paste mechanism when mobilizing, increasing their numbers within the host genome as a result. 8 RTE-specific mobilization encompasses an RNA intermediate that is reverse transcribed into a complementary DNA (cDNA) “copy,” which is then inserted into the genome at a locus different to that of the source element. 9 Within the human genome, the only known TEs to be currently active are the non-long terminal repeat (non-LTR) retrotransposons consisting of long interspersed elements (LINEs), short interspersed elements (SINEs) and SINE-VNTR (variable number tandem repeat)-Alus (SVAs). 10 SVAs, the youngest of the RTEs in the human genome and hominid-specific, are so termed after their composite domains. 11 SVAs consist of a hexamer repeat (CCCTCT), Alu-like sequence on the antisense strand, GC-rich VNTR, SINE region, a poly A-tail and target site duplications flanking the site of insertion (Figure 1(a)).12–14
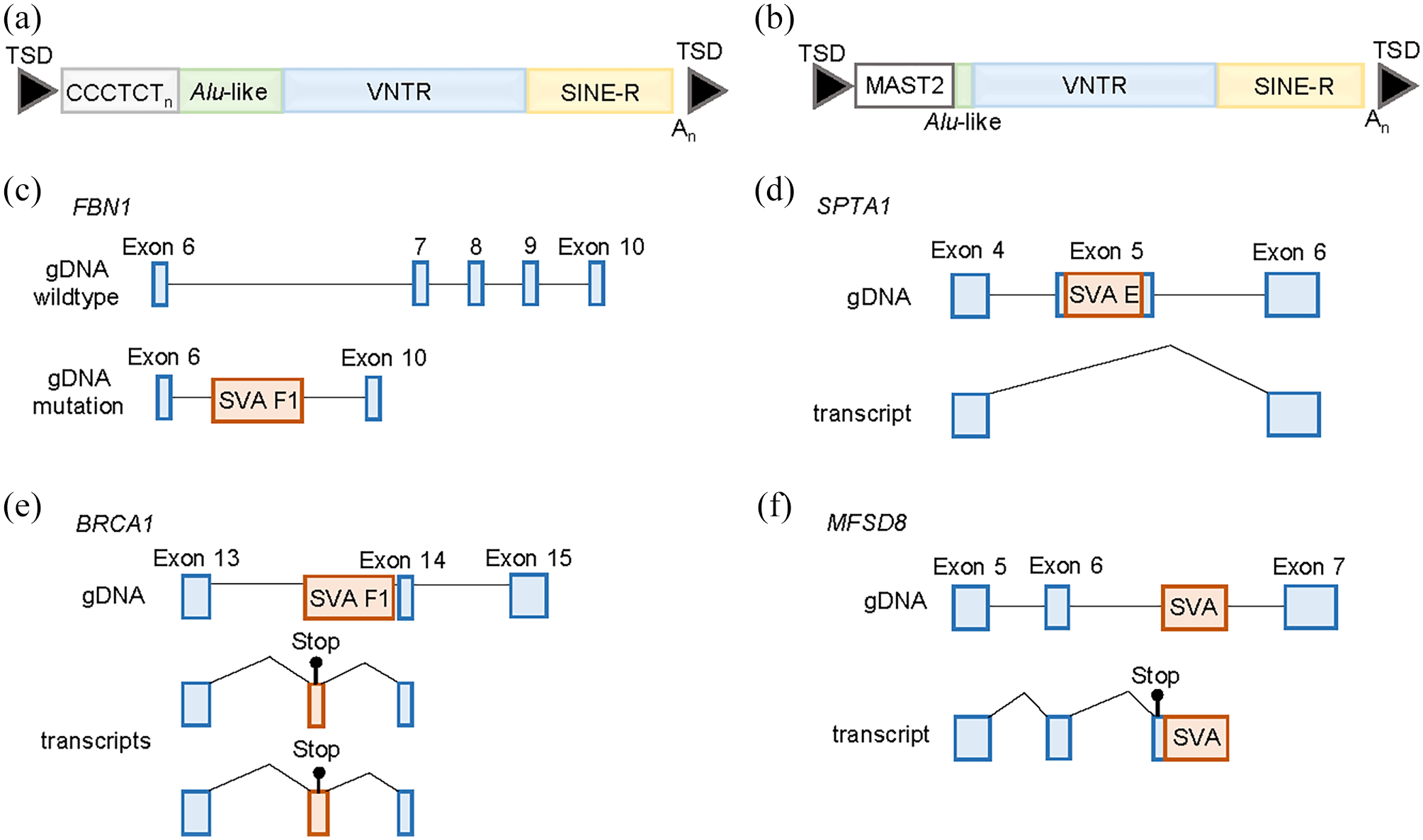
SVA structure and mechanisms through which they are associated with disease. (a) A complete SVA consists of a hexamer repeat (CCCTCT) variable in length, an Alu-like sequence on the antisense strand, a variable number tandem repeat (VNTR), a SINE-R domain, a poly A-tail and are usually flanked by target site duplications (TSDs). (b) An SVA F1 consists of the VNTR, SINE-R, and poly A-tail but is lacking the CCCTCT domain and the majority of the Alu-like sequence. At the 5′ end is sequence from the MAST2 exon 1 that can vary in size. (c) An example of a genomic deletion upon SVA insertion where a region of ~36.8 kb is deleted that includes exons 7-9 of the FBN1 gene. (d) Exon skipping induced by the insertion of an SVA into exon 5 of the SPTA1 gene resulting in an inframe deletion and the production of an abnormal protein. This was associated with hereditary elliptocytosis and hereditary pyropoikilocytosis. (e) Exonization of an intronic SVA insertion that introduced a premature stop codon in the BRCA1 gene in a family with early onset breast cancer. (f) An SVA insertion into an intron of the MFSD8 gene that activates existing cryptic splice sites causing missplicing and the introduction of a premature stop codon in the transcript. This was associated with neuronal ceroid lipofuscinosis 7. (A color version of this figure is available in the online journal.)
LINE-1 (L1) elements are the only active autonomous RTE in humans. 10 Encoding the proteins required to retrotranspose, L1s are able to mobilize themselves 15 and other RTEs such as Alus 16 and SVAs.11,17–19 L1s retrotranspose by way of target primed reverse transcription (TPRT), a process that begins when L1 RNA is transcribed by RNA polymerase II before being exported to the cytoplasm of the cell. 20 Within the cytoplasm the two open reading frames, ORF1 and ORF2, encoded by the L1 will be translated into proteins, ORF1p and ORF2p respectively. 21 ORF1p binds to nucleic acids, 22 while ORF2p has both reverse-transcriptase and endonuclease activities.23,24 Following translation in the cytoplasm, ORF1p and ORF2p join L1 RNA to form a ribonucleoprotein complex (L1 RNP) which is then transported back into the nucleus.25,26 To ensure a functional L1 is more likely to be inserted into the host genome the L1 encoded proteins demonstrate a cis preference for their encoding RNA. 27 Facilitated by its endonuclease activity, ORF2p nicks the bottom strand of the hosts DNA at the 5′TTTTAA3′ consensus sequence at the TA site. Primed by a 3′-hydroxyl group liberated at the TA site, ORFp2 then reverse transcribes the L1 RNA into cDNA before nicking the top strand of the host DNA to allow integration of the cDNA into the genome. 25 Finally, the complementary strand of DNA is synthesized. 20
SVAs are divided into subtypes (A–F) according to the SINE region, with the oldest elements belonging to A and F being the youngest. 11 A subsequent seventh subtype was identified, SVA F1, containing a 5′ transduction of the sequence from the MAST2 gene and the incorporated MAST2 sequence having been shown to act as a positive regulator of transcription (Figure 1(b)).28–31 Elements from the subtypes D, E, and F1 were found to retrotranspose to varying degrees in multiple cell lines.17,19 These studies demonstrated that ORF2p was required for retrotransposition, while the need for ORF1p depended on the SVA element co-transfected with the L1 driver construct.17,19 This may be due to size or sequence differences of the SVAs tested. L1 mobilization requires both proteins; however, Alu retrotransposition only requires ORF2p. 16 The rate of retrotransposition in the population for Alu, L1, and SVAs had been estimated as 1/21, 1/212, and 1/916 live births, respectively. 32 However, using a pedigree analysis, the rate was reported as 1/40 for Alus and 1/63 for both L1s and SVAs. 33 The rate of SVA mobilization was much higher in the pedigree analysis than the previous estimate.
In the literature, 24 SVA insertions were identified as being associated with disease. The mechanisms behind this fell into three broad categories of large genomic deletions upon insertion of an SVA, exonic insertions leading to loss-of-function mutations, and those causing aberrant splicing (Table 1). For the 21 SVAs that the subtype was reported, they all belonged to the three most recently active subtypes (E, F, and F1), which are human specific. The first disease-causing SVA was identified in 1994 and 12 such insertions had been identified up until 2016. 2 In the past 6 years, this number has doubled as technological advances have allowed the analysis of these types of elements more widely. The affordability of high-depth whole-genome sequencing and a range of bioinformatics tools developed to call retrotransposon insertions from these data have been an important advance in characterizing this type of variation genome-wide. 34 It has also enabled population scale analyses to identify polymorphic insertions and provide a reference of variants common to a population for comparison.3,4 Here we discuss the disease-associated SVAs and the mechanisms through which they act to provide insight into how these elements influence genomic function.
Disease-associated SVAs.
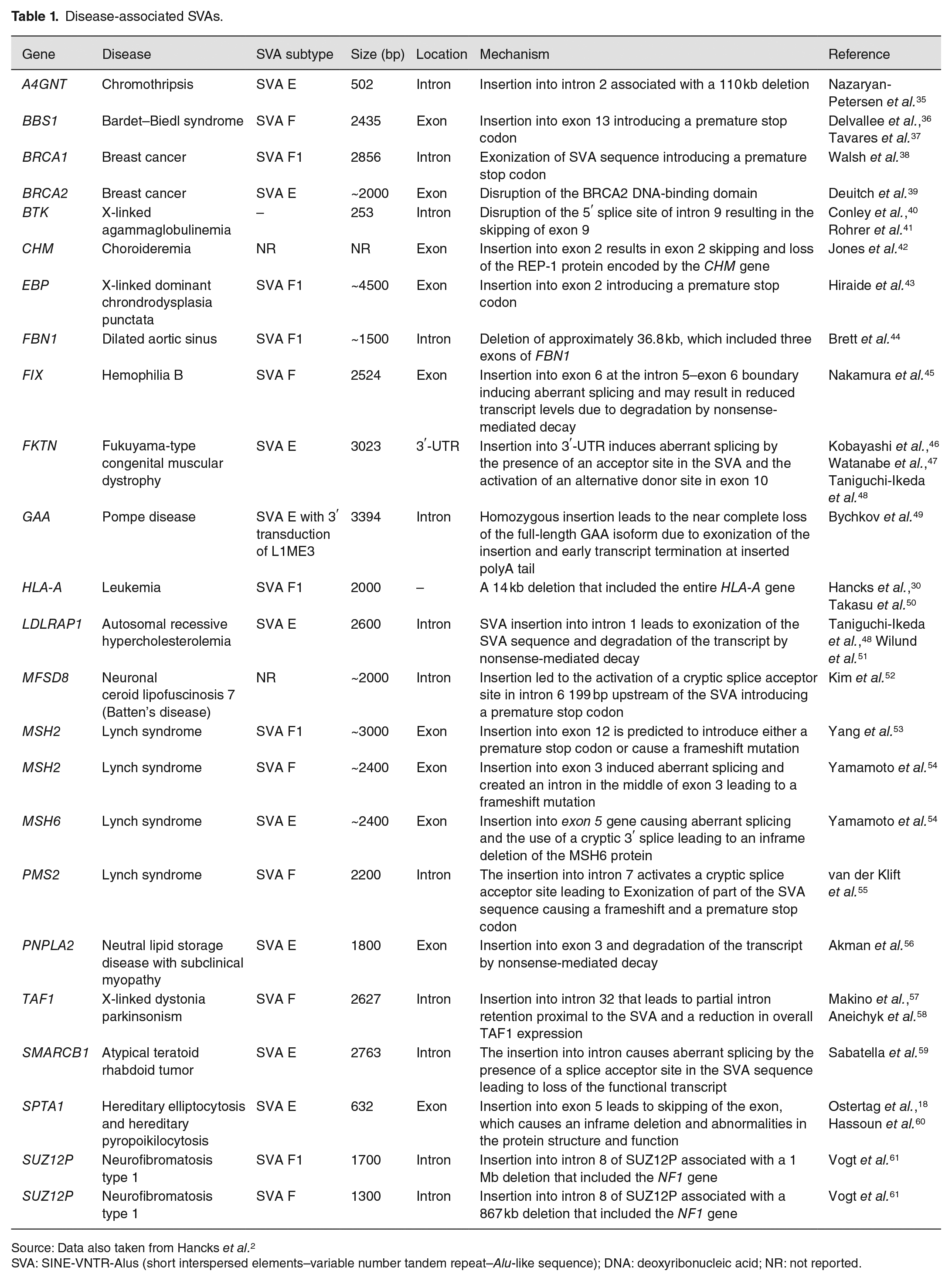
Source: Data also taken from Hancks et al. 2
SVA: SINE-VNTR-Alus (short interspersed elements–variable number tandem repeat–Alu-like sequence); DNA: deoxyribonucleic acid; NR: not reported.
Exonic SVA insertions and loss-of-function mutations
SVAs contain stop codons in their sequence and insertions into exons will often introduce a premature stop codon or cause a frameshift mutation, leading to the transcript being degraded by nonsense-mediated decay. Several examples of SVAs causing loss-of-function mutations via exonic insertion have been reported (Table 1). These include an SVA F in the PNPLA2 gene associated with neutral lipid storage disease with subclinical myopathy, an SVA E in the BRCA2 gene associated with breast cancer and an SVA F1 in the MSH2 gene associated with the cancer predisposition disease Lynch syndrome.39,53,56 An SVA F insertion in exon 13 of the BBS1 gene was identified as the second most common pathogenic variant associated with the rare recessive disease Bardet–Beidl syndrome, which occurred in a common ancestor at an estimated 74 generations ago.36,37 In addition, a recent report of SVA F1 insertion with accompanying 5′ and 3′ transductions of Alu sequences was associated with X-linked dominant chondrodysplasia punctata (CDPX2). CDPX2 occurs almost exclusively in females and is associated with skin, bone, and eye abnormalities caused by pathogenic variants in the EBP gene. 43 Structural variant analysis of whole-genome sequencing data identified the SVA F1 in exon 2 of the EBP gene in the proband and her affected mother and inspection of the SVA F1 sequence provides evidence for the introduction of a premature stop codon. 43
SVA insertions associated with large genomic deletions
A deletion of three FBN1 exons (7–9) from intron 6 to intron 9, spanning approximately 38.6 kb at 15q21.1 with a concomitant SVA F1 insertion, was identified in a study of a child with mildly dilated aortic sinus (Figure 1(c)). 44 Mutations in the FBN1 gene typically result in an autosomal dominant connective tissue disorder termed Marfan syndrome (MFS), which affects the skeletal, cardiovascular, and ocular systems. 62 In this case, the child displayed no other features indicative of an MFS diagnosis. Loss of exons 7 and 8 likely influence correct folding of the fibrillin protein,63,64 while additional disruption of the coding sequence of the messenger RNA (mRNA) may result from the acceptor and donor splice sites contained within the inserted SVA. Large intragenic deletions and chromosomal imbalances are only responsible for an estimated 1–2% of MFS cases,65,66 and no previously reported cases involved the deletion of exons 7–9.
Chromothripsis (CTH) is the occurrence of hundreds of DNA double-stranded breaks (DSBs) typically within small genomic regions, though regions as large as entire chromosomes have been identified.67,68 Initially, CTH was exclusively associated with cancer pathogenesis; however, when later identified in some congenital and developmental disorders, limited cases of germline-CTH (GCTH) were described.69–71 The DSB are typically repaired to generate genomic fragment rearrangements involving translocations, inversions, and insertions. With few resultant deletions being observed, CTH is relatively balanced. 70 In a case of familial G-CTH, numerous break points were identified in chromosome 3 accompanied by a 502 bp 5′-truncated SVA E element insertion into intron 2 of the A4GNT gene. 35 Associated with this insertion was a 110 kb deletion at the 5′-end of the insertion. The insertion location within a sequence reminiscent of the L1 endonuclease cleavage site and truncation of the insertion itself at its 5′-end suggest L1-mediated retrotransposition as the mechanism by which this insertion occurred.18,23 In addition, the location of the deleted sequence at the 5′-end of the insertion and lack of target-site duplications, are features reminiscent of previously described retrotransposon insertions with concomitant deletion events.72,73 Finally, the 100% sequence match between the inserted SVA E element and its source element, an SVA E located on chromosome 7, further indicates retrotransposition as the mechanism of this insertion event.
The insertion and deletion events most likely occurred concomitantly due to several factors. The authors suggest that ORF2p endonuclease activity precipitated multiple DNA breaks leading to G-CTH and mediated SVA E retrotransposition associated with a large-scale deletion. Specifically, the authors postulate a model whereby two mispaired AluSx elements flanking the deleted segment, both in the same orientation, may have predisposed the segment for deletion, mediating chromatin looping and arranging the segment proximally to ORF2p. This facilitated cleavage and SVA retrotransposition.
When investigating genetic samples provided for bone marrow testing individuals from three apparently unrelated Japanese families, another study found SVA retrotransposition accompanied by a large 14 kb deletion comprising the entire HLA-A gene. 50 The 2 kb insertion SVA was later identified as belonging to the SVA F1 subfamily. 30 Interestingly, though the three families were apparently unrelated, they shared identical HLA-A haplotypes and each family originated from the same area of Japan, North Kanto. This suggested a possible common ancestor from which the original deletion and insertion was inherited as a founder mutation. 50
Accounting for approximately 8–10% of large NF1 deletions, “atypical” deletions have non-recurrent breakpoints and vary by size and the number of genes within the deleted region.74,75 Prior analysis of the few highly characterized atypical NF1 deletions suggested they were due to non-homologous end joining (NHEJ). Two instances of large atypical NF1 deletions within SUZ12P intron 8 in unrelated patients were found to be accompanied by SVA retrotransposition at the deletion breakpoints. 61 One patient possessed a 1 Mb deletion with a 1.7 kb SVA insertion likely sourced from one of the most active SVA elements and belonging to the SVA F1 subfamily, H10_1 on chromosome 10q24.2.29,30 The other patient displayed a 867 kb deletion accompanied by an SVA insertion highly homologous to SVA F element H6_1084 on chromosome 6q22.31. The sites of both insertions were separated by 3067 bp, highlighting the concept of retrotransposition hotspots within the human genome as postulated by earlier studies.40,76–78 The study concluded that the origin deletion and insertion events occurred concomitantly during early post-zygotic development for each case (indicated by the somatic mosaicism present in one patient and grandmother of the other). Endonuclease cleavage sites located within SUZ12P intron 8 and long polyT tracts at integration sites suggested the insertion events were mediated by L1-associated TPRT. Again, it was postulated that SVA insertion resulted in loop-like conformational changes to chromatin. This conformational change brought the SUZ12P sequence ending with a newly inserted SVA element into proximity of the telomeric NF1 gene region, facilitating their ligation most likely by NHEJ. 79
SVA-induced aberrant splicing
More than half of the disease associated SVA insertions outlined in Table 1 cause aberrant splicing patterns, that include exon skipping, activation of cryptic splice sites up or downstream of the SVA, and the inclusion of the SVA sequence in gene transcripts. This can result in smaller proteins that can impair their normal function or frameshift mutations and nonsense-mediated decay leading to disease. Although, not in all cases can the precise effects be determined. For example, an SVA insertion into exon 6 of the FIX gene that causes Hemophilia B alters normal splicing of exons 5 and 6; however, whether this was due to exon skipping or exonization was unknown. 45
Exon skipping
The skipping of exons due to mutation can lead to disease either through altering the reading frame or protein structure. SVA insertions in exons or at the exon–intron boundary have led to three cases of disease (Table 1). An intronic insertion, close to the exon–intron boundary, in the BTK gene caused skipping of exon 9 by disrupting the 5′ splice site of intron 9 resulting in an inframe deletion, an unstable protein, and the immune system affecting disorder X-linked agammaglobulinemia.40,41 The insertion of an SVA into exon 5 of the SPTA1 gene causes exon 5 skipping that leads to an inframe deletion producing abnormalities in the protein structure and function and is associated with the heterogeneous red blood cell disorder hereditary elliptocytosis and hereditary pyropoikilocytosis (Figure 1(e)).18,60 The third example of SVA-induced skipping is an insertion in exon 2 of the CHM gene and is associated with choroideremia, a condition characterized by progressive vision loss. 42 This insertion causes exon 2 skipping and an absence of the REP-1 protein encoded by the CHM gene.
Exonization of SVA sequence
An SVA in the sense orientation contains multiple cryptic splice sites throughout its sequence and several instances of these SVA splice sites being used have been reported. 30 In addition, the most recent subfamily of SVAs (SVA F1) was created when exon 1 of the MAST2 gene was spliced into the Alu-like region of an SVA F and was subsequently retrotransposed (Figure 1(b)).28–30 This subfamily rapidly expanded with more than 80 SVA F1s in the human genome 29 and six disease-associated insertions (Table 1). The SVA sequence also contains multiple stop codons within its sequence; therefore, exonization of the SVA is likely to introduce premature stop codons causing nonsense-mediated decay or produce truncated proteins. Six of the disease-associated SVAs reported in Table 1 act through this mechanism, five of which are located within introns and one in a 3′-UTR. The SVA in the 3′ UTR of the FKTN gene is a founder insertion and is associated with Fukuyama-type congenital muscular dystrophy (FCMD).46,47 FCMD is a type of muscular dystrophy accompanied by abnormalities in the brain and eyes and is predominantly found in Japan. The insertion causes an abnormal splicing event with a rare alternative donor site in exon 10 (last exon) and an acceptor site in the SVA being used. This leads to a truncation of the normal fukutin protein, and the final 129 amino acids being coded for by the SVA causing mislocalization of the protein. 48
An SVA in intron 1 of the LDLRAP1 gene is associated with autosomal recessive hypercholesterolemia leading to no detectable LDLRAP1 mRNA in patient cells due to abnormal splicing and degradation of the SVA-containing transcript.48,51 Three of the intronic SVA insertions that undergo exonization are in genes in which a germline mutation predisposes an individual to certain cancers. A case of Lynch syndrome was associated with an SVA in intron 7 of the PMS2 gene that led to the exonization of 71 bp sequence consisting of nucleotides from the target site duplication and the SVA itself. 55 The remainder of the SVA sequence was spliced out using a cryptic donor site in the SVA sequence and the canonical splice acceptor of intron 7. The inclusion of this sequence caused a frameshift, which introduced a stop codon and the transcript being degraded. Using a long-read sequencing approach to analyze the DNA of a family affected by early-onset breast cancer, an SVA insertion was identified in intron 13 of the BRCA1 gene in the proband, which segregated with breast cancer in the family. 38 Previous panel testing and exome sequencing had been unable to identify the genetic variant involved. The SVA resulted in two additional transcripts, depending on the size of the SVA sequence included, to be expressed both of which contained premature stop codons (Figure 1(d)). Atypical teratoid rhabdoid tumor is a rare and aggressive pediatric tumor caused by the biallelic inactivation of SMARCB1 with nearly one-third of cases carrying a predisposing germline variant. One such germline variant was identified as an SVA in a pair of siblings diagnosed with the disease. 59 The SVA was located in intron 2 of the gene, which caused splicing from exon 2 into the SVA using a splice acceptor in the Alu-like region. This is likely a highly penetrant variant due to both siblings being affected, and the insertion only present in the mother in a mosaic state. 59 The final example of an exonized SVA in disease is located in the GAA gene and is associated with Pompe disease, an autosomal recessive lysosomal storage disorder. 49 The patient was homozygous for an insertion in intron 15 of GAA gene, which caused exon 15 to be spliced into the SVA and termination of the transcript at the insertion’s poly A-tail and the almost complete absence of the full-length isoform.
Activation of cryptic splice sites
SVAs have been shown to alter splicing by activating existing cryptic splice sites at the locus in which they insert, in both introns and exons. An SVA F insertion in exon 3 of the MSH2 gene in a patient with Lynch Syndrome led to the use of a new donor and acceptor splice site up and downstream of the SVA insertion. This divided exon 3 into two, creating an intron in the middle of the existing exon. 54 This resulted in shorter mRNA and a frameshift mutation. A second case of Lynch Syndrome was associated with an SVA E insertion into exon 5 of the MSH6 gene, also through changes to normal splicing patterns. 54 This insertion altered the splice acceptor site that was used to downstream of the SVA causing an inframe deletion in exon 5.
An SVA in intron 32 of the TAF1 gene was associated with X-linked dystonia-parkinsonism (XDP). 57 The presence of the SVA was associated with reduced TAF1 expression and the inclusion of a cryptic exon in intron 32 5′ of the SVA. Removal of the SVA using CRISPR/Cas9 rescued this aberrant transcription normalizing TAF1 expression. 58 In addition to the presence of the SVA influencing XDP, the length of the CCCTCT domain at the 5′-end of the SVA was associated with age of disease onset. 80 The median repeat number was found to be higher in the basal ganglia and cerebellum compared to the blood of the same individual suggesting somatic repeat instability is a feature of specific brain regions and could play a role in disease manifestation. 81 Therefore, size variation of the SVA should also be an important factor when evaluating their functional impact.
Whole-genome sequencing was performed on a patient diagnosed with neuronal ceroid lipofuscinosis 7 (CLN7), a form of Batten’s disease, who was heterozygous for a known pathogenic mutation in the MFSD8 gene to identify the second mutation involved. An SVA in intron 6 of the gene was identified, which was absent from over 800 whole genomes and present in the mother of the proband. Analysis of RNA from the patient demonstrated missplicing of exon 6 into a cryptic splice acceptor site 199 bp upstream of the SVA introducing a stop codon and was predicted to lead to premature termination of translation (Figure 1(g)).
Modulation of SVA-induced aberrant splicing using antisense oligonucleotides
Antisense oligonucleotides (AOs) are single-stranded synthetic nucleic acid analogues that can be designed to target specific sequences to modulate gene expression and several have been licensed to treat diseases such as Duchenne muscular dystrophy and spinal muscular atrophy. 82 AOs have been designed to target missplicing caused by two SVA insertions, one of which (milasen) was approved by the Food and Drug Administration and expedited institutional review board to be administered by an intrathecal bolus injection to the patient it was designed for in a “n-of-1” situation.48,52,83
AOs were designed to restore translation of the full-length fukutin protein to correct the missplicing caused by the SVA insertion in the 3′ UTR of the FTKN gene that causes FCMD. The AOs targeted acceptor, donor, and exonic splicing enhancer sites and a cocktail of three AOs achieved the greatest recovery of FKTN mRNA in lymphoblast and myotube patient cell lines. 48 The AO cocktail was also tested in mice rescuing the full-length FKTN mRNA and restoring the normal protein. 48 This provides a potential treatment for FCMD patients through the modulation of splicing.
One of the pathogenic mutations in a single patient with the fatal neurodegenerative disorder neuronal ceroid lipofuscinosis 7 (CLN7) was identified to be an SVA insertion that altered normal splicing of the MFSD8 gene and splice-modulating AOs were designed to correct this. The AOs targeted the cryptic splice acceptor site activated by the SVA insertion and nearby splicing enhancers. The lead candidate was chosen and was named milasen. 52 Milasen was tested in patient fibroblasts more than tripling the amount of normal splicing and alleviated cellular phenotypes associated with lysosomal dysfunction. Due to deterioration of the patient, expedited approval was given for use of the AO and treatment with milasen resulted in the reduction and frequency of seizures experienced by the patient. 52 This demonstrates the rapid development of a targeted personalized therapeutic, and that SVAs, which alter normal splicing, may be amenable to modulation using AOs.
Common SVA insertions and disease risk
The insertions discussed so far are rare and cause a robust phenotype leading to disease. However, common insertions could influence disease risk with more subtle functional effects. For example, an SVA in intron 8 of the CASP8 gene is a common polymorphic element that was associated with the retention of intron 8, an increased risk of breast cancer and protective for prostate cancer. 84 This polymorphic SVA is in strong linkage disequilibrium (LD) with single nucleotide polymorphisms (SNPs) that were identified as risk variants in genome-wide association studies (GWAS). 84 This approach of LD analysis of SVA polymorphisms and known GWAS risk variants would be useful to identify those SVAs that could be the causative variant at known disease-associated loci. A focused analysis of reference SVA polymorphisms and Parkinson’s disease (PD)-associated SNPs identified an SVA on chromosome 17 in strong LD with a known PD risk variant. 85 A genome-wide approach across multiple diseases, similar to analysis performed for Alu variation, 86 could provide insight into common SVA variation and disease risk.
Conclusions
The list of SVA insertions associated with disease is increasing and as the number of genomes analyzed for complex structural variants expands it is likely that the number of disease-causing SVAs will as well. Understanding the mechanisms through which these elements cause disease not only contributes to the overall knowledge regarding their function but also to potential novel therapeutics that modify their effects.
Footnotes
Authors’ contributions
ALP and LMS reviewed the literature, ALP generated the table and figure and all authors contributed to the writing and editing of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: ALP, LMS, and SK are funded by Multiple Sclerosis Society of Western Australia.