
Abstract
Cancer-associated cachexia (CC) is a pathological condition characterized by sarcopenia, adipose tissue depletion, and progressive weight loss. CC is driven by multiple factors such as anorexia, excessive catabolism, elevated energy expenditure by growing tumor mass, and inflammatory mediators released by cancer cells and surrounding tissues. In addition, endocrine system, systemic metabolism, and central nervous system (CNS) perturbations in combination with cachexia mediators elicit exponential elevation in catabolism and reduced anabolism in skeletal muscle, adipose tissue, and cardiac muscle. At the molecular level, mechanisms of CC include inflammation, reduced protein synthesis, and lipogenesis, elevated proteolysis and lipolysis along with aggravated toxicity and complications of chemotherapy. Furthermore, CC is remarkably associated with intolerance to anti-neoplastic therapy, poor prognosis, and increased mortality with no established standard therapy. In this context, we discuss the spatio-temporal changes occurring in the various stages of CC and highlight the imbalance of host metabolism. We provide how multiple factors such as proteasomal pathways, inflammatory mediators, lipid and protein catabolism, glucocorticoids, and in-depth mechanisms of interplay between inflammatory molecules and CNS can trigger and amplify the cachectic processes. Finally, we highlight current diagnostic approaches and promising therapeutic interventions for CC.
Impact statement
Cancer cachexia is a sequela of catabolism leading to poor muscle strength, reduced locomotion, weight loss, and intolerance to therapy. Greater than 80% of advanced cancer patients display cachexia, and it is a major cause of cancer-related deaths. Cancer cells harbor host tissue metabolism by secretory molecules and cargo packaged extracellular vesicles (EVs), and skew the entire host metabolism toward catabolism while themselves being anabolic. Currently, there is no standard therapy available that can effectively reverse cancer-related cachexia. Therefore, robust preclinical research in the context of etiology, initiation, progression, and therapy followed by robust clinical validation are warranted. In this regard, our present review discusses the imbalance of host metabolism, early changes during precachexia, and highlights the molecular pathways. In addition, we shed a light upon current diagnostic and therapeutic challenges associated with cancer-associated cachexia.
Introduction
Reprogramming of metabolism is one of the crucial hallmarks of cancer. 1 Elevated glycolysis, hepatic gluconeogenesis, lipolysis, and proteolysis of skeletal muscles result in reduced body fat and progressive weight loss in cancer patients. 1 This systemic tissue wasting sequela termed CC diminishes locomotion leading to poor quality of life, poor prognosis, and increased mortality. More than 80% of advanced-stage cancer patients display cachexia, and it accounts for about 25% of cancer-related deaths.2,3 Nearly 30% of the CC patients have associated cardiac disorders with the risk of developing cardiac cachexia. 4 Cachexia might be present in the early-stage development of cancer even before the onset of signs and symptoms of malignancy. 2 Initially, cachexia was thought to be a muscle protein and fat depletion metabolic disorder.5,6 However, recent studies have established that cachexia is a multifactorial pathological condition associated with hypoxia, tissue pH, anorexia, altered metabolism, and chronic inflammation.6,7 Hypoxia-inducible factor 1-α (HIF-1α) and hypoxia-inducible factor 2-α (HIF-2α) have been revealed to stimulate CC through diverse mechanisms comprising loss of appetite, activation of phosphoenolpyruvate carboxykinase (PEPCK), and the Cori cycle.8,9 Indeed, pro-inflammatory cytokines such as tumor necrosis factor–alpha (TNF-α) and interleukin-6 (IL-6) were shown to be elevated in various CC in vitro and in vivo models and are proposed to be diagnostic markers of CC.8,10–12 Studies conducted by Oliff et al. 13 and Baltgalvis et al. 14 demonstrated that supraphysiological IL-6 and TNF-α levels led to early CC and deaths in animal models. So far, monotherapies targeting numerous inflammatory cytokines have failed to efficaciously treat CC, owing to its multifactorial landscape.15–17 Two independent studies by Stovroff et al. 18 and Vaisman and Hahn 19 demonstrated that TNF-α induces corticotrophin-releasing hormone, and it increases the firing of glucose-sensitive neurons leading to anorexia followed by cachexia. Furthermore, the cytokine surge is known to induce depression-induced anorexia, lipolysis via Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3), and Nuclear factor kappa- light chain enhancer of activated B cells (NF-κB) pathways and β-adrenergic activation connect CC with the central nervous system (CNS).20,21 Nevertheless, loss of appetite is not a primary cause of cachexia during cancer progression, as the accompanying engrossment of massive skeletal muscle wasting does not occur during anorexia, and nutritional supplementation fails to impede loss of lean body mass.22–24 Currently, the precise mechanisms that promote and govern CC are yet to be deduced, thereby limiting the therapeutic targets for the treatment of CC. Therefore, the current review provides in-depth information about spatio-temporal changes in tissue microenvironment and systemic metabolism during the initial stages of CC development. We also focus on the energetics of the cancer cell and surrounding stromal microenvironment, rate-limiting steps, and the mechanism of absconding checkpoints. Furthermore, we provide a comprehensive overview of molecular insights into tumor cell anabolism and normal tissue catabolism status during progressive CC. Finally, we highlight the current difficulties and complications in the early diagnosis and treatment of CC.
Spatio-temporal changes during cachexia progression in cancer patients
CC is divided into three successive clinical stages such as precachexia, cachexia, and refractory cachexia (RC), although not all the patients progress through this continuum.21,22 The degree of clinical manifestations and the condition of patients, including locomotory index and probable survival period, are taken into account while staging the disease. 3 The staging of CC has crucial implications for management and treatment which go beyond merely an understanding of alleviating symptoms.
Cancer precachectic state
Precachexia is associated with less than 5% of unintentional total body weight loss in 6 months. Cancer precachectic state (CPC) is accompanied by underlying chronic disease, inflammation, loss of appetite, and/or metabolic alterations. 3 Notably, CPC patients display heterogeneity, with some progressing rapidly, while others remain weight stable. In all preclinical and clinical settings, CPC displays systemic involvement of inflammation with elevated levels of TNF-α and IL-6, insulin resistance, in addition to sarcopenia and weight loss. 25 An important distinction between cachexia and anorexia is that weight loss during cachexia is not solely confined to reduced calorie intake. Indeed, it is noteworthy that not all the malnourished patients are cachectic, while all cachectic patients are malnourished.3,26 Malnutrition during cancer progression can even be due to the direct impact of tumors on the gastrointestinal tract, therapy-induced nausea, satiety, and impaired appetite regulation. The current understanding of metabolic balance and body weight is largely derived from the integrative physiological research into obesity and its associated etiopathologies. 27 The interplay of chronic inflammation and immune system modulation with dysregulated metabolism in obesity extensively involves inflammatory mediators that overlap with potential molecular drivers of CPC, suggesting the involvement of similar flawed signaling pathways, albeit with dramatically contradicting end-stage clinical manifestations.28,29 Higher glucose demands are met in the initial stages of CC by glycogenolysis and reduced glycogen storage in muscle, and preceding massive gluconeogenesis.30,31 During CPC, muscle protein synthesis and degradation have been found to be normal, with normal atrogens, while myogenesis and muscle contractility have been found to be reduced.30,31 Oxidative stress, inflammatory mediators, and reactive oxygen species (ROS) emission have been found to be elevated with unaltered autophagy and mitophagy.32,33 Similarly, elevated mitochondrial ROS generation and oxidative stress have been shown to precede terminal cachexia development in tumor-bearing mice. 34
Cachexia
Cachexia is diagnosed by >5% unintentional total body weight loss over the previous 6 months despite the normal nutritional food intake, or the combination of >2% ongoing weight loss with body mass index (BMI) < 20 or sarcopenia. 3
Protein homeostasis in skeletal muscle is imbalanced toward decreased protein synthesis and elevated breakdown in response to cancer.30,31 The hyperactivated ubiquitin-proteasome and autophagy pathways are primarily responsible for this unevenness.30,31 Transcriptional upregulation of numerous E3 ligases, including muscle-specific RING finger protein 1 (MURF1), muscle atrophy F-box protein (MAFBX), FBXO30, and FBXO31, and increased turnover of myofibrillar proteins result in muscle atrophy during CC.35–37 These amino acids are directed mainly toward gluconeogenesis to meet the requirements of growing tumor mass. Furthermore, patients with CC often display insulin resistance. Under normal physiology, insulin regulates carbohydrate metabolism and muscle protein balance to maintain blood glucose levels.38,39 Insulin resistance mobilizes proteins from muscle to liver through the suppression of the PI3K-Akt pathway and by upregulation of the proteasomal pathway.40,41 These amino acids released from muscle protein turnover are directed to the liver for gluconeogenesis as an alternate source of energy production during the period of glucose scarcity. 42 These dysregulated energy-cumbersome metabolic activities in the cancer cells can henceforth withstand energy depletion, necessitating more muscle breakdown as cancer progresses.
Furthermore, increased lipolysis leading to elevated levels of fatty acids or glycerol has been reported to fuel liver gluconeogenesis and insulin resistance in CC patients. Insulin resistance during CC dysregulates the PI3K/Akt/mTOR pathway in adipose tissues resulting in increased adipose tissue wasting. 43 Altogether, the complete metabolism of cancer cells concentrates on anabolism while the surrounding host tissues and systemic metabolism of the host under continuous catabolism lead to profound muscle and weight loss in CC patients.
RC
RC is notable for its poor World Health Organization (WHO) physical performance score and survival expectancy of <3 months, and these patients are suited only for psychosocial support and palliative care. 3 The severity of cachexia increases as the disease progresses to metastasize. Bodyweight and circulating albumin (ALB) levels of patients with RC were significantly lower compared to patients with CPC while C-reactive protein (CRP) was found to be profoundly increased in these patients.44,45 Recently, Suno et al. 46 demonstrated that increased plasma fentanyl levels could be the biomarker for RC. In this study, the authors analyzed the fentanyl levels and hence the exclusion criteria of the patients selected in the study was use of drugs, as supportive therapy, that might affect the metabolism of CYP3A4, and antipsychotic drugs. Suno and co-workers have shown the remarkable association of dose-adjusted fentanyl concentrations with refractory cancer cachexia. RC is significantly associated with cardiac atrophy, arrhythmias, and electrolyte imbalance that increase the risk of thromboembolic events, cardiac arrest, breathing difficulties, aspiration pneumonia, swallowing difficulties, reduced joint movements, gastrointestinal muscle atrophy, poor wound healing, and sepsis. Not surprisingly, clinical data showed high mortality within a few weeks of the development of RC. 47
The detailed differences between CPC, CC, and RC are summarized in Table 1.14,30,31,35,37,44,46,48–73
Characterization of different phases of cachexia.
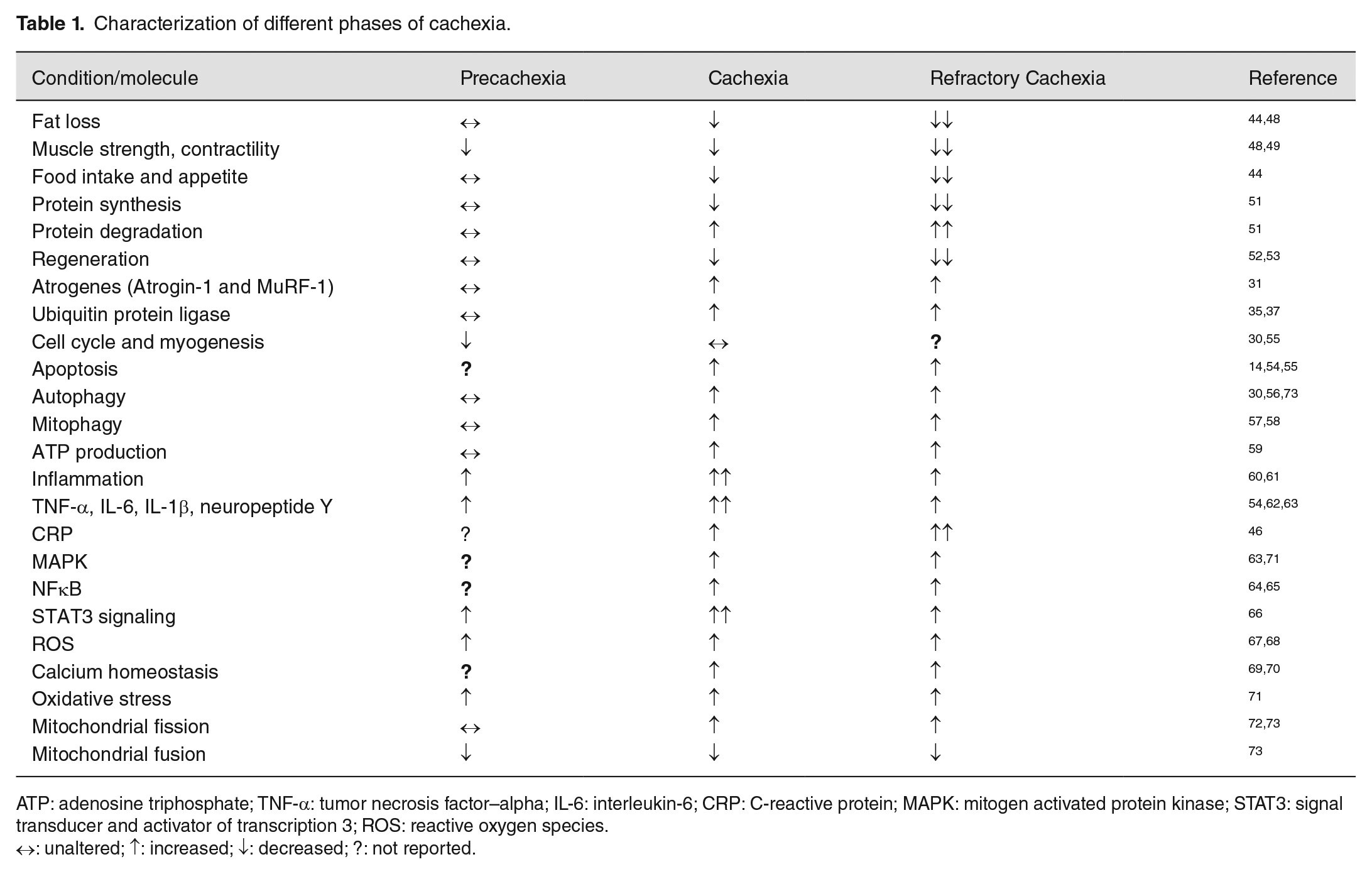
ATP: adenosine triphosphate; TNF-α: tumor necrosis factor–alpha; IL-6: interleukin-6; CRP: C-reactive protein; MAPK: mitogen activated protein kinase; STAT3: signal transducer and activator of transcription 3; ROS: reactive oxygen species.
↔: unaltered; ↑: increased; ↓: decreased; ?: not reported.
Energetics of cancer cells and host tissues
Increased catabolism is observed in cancer patients leading to unsustainable muscle and fat loss, thereby causing high morbidity and mortality.7,74 The factors such as old age, tumor progression, comorbid conditions, nutritional deficiency, drugs, and medical interventions were found to be the major determinants of the resting energy expenditure (REE). 75 Tumors demand a continuous supply of glucose and energy for their uninterrupted growth and proliferation and alter the energy balance of host tissue by eliciting inflammatory pathways which augment both systemic and locally mediated catabolic events. 76 Tumors also consume the increased amount of macronutrients directly. When tumors reach the size >0.75 kg, the energy consumption by the tumor is quantitatively important. 74 The average daily energy expenditure of cancer patients is between 1600 and 1800 kCal. 77 Lieffers et al. 78 demonstrated that in metastatic colorectal cancer patients, there is a cumulative increment in REE which accounts for about ~17,700 kCal over 3 months, and this high demand of REE itself can contribute to a substantial reduction in body weight. In this study, the authors have tried to link the REE to energetically demanding tissues such as liver, spleen, skeletal muscles, adipose tissues, and tumor mass with advanced cancer and cancer cachexia–associated weight loss. Another mechanism that amplifies catabolic signals while desensitizing muscles to anabolic signals is physical inactivity; for instance, Kortebein et al. 79 reported that bed rest for 10 days causes a 6% loss in lower limb muscle, a 30% drop in muscle protein synthesis, and a 16% loss in isokinetic muscle strength in otherwise healthy adults over 65 years of age. Bed rest elevates catabolic responsiveness of the muscles to cortisol by threefold. 80 Muscle anabolism was induced by physical exercise in these patients and though fatigue was not reduced, quality of life was improved. 81 Although bed rest in healthy elderly subjects leads to signs and symptoms of disuse atrophy, more studies are needed to understand the molecular mechanisms governing disuse atrophy and CC, and exploring whether they are similar or not.
Imbalance of anabolism and catabolism: insight into the molecular level
Our present understanding of the basic mechanisms that promote CC is based on three lines of experimental evidence. First, Ni et al. 82 demonstrated that surgical removal of cachexia-related tumors entirely (when practicable) could reverse CC in pancreatic xenograft models. Tumors are thus required not only for the induction of cachexia but also for its maintenance. Second, Norton et al. 83 showed pro-cachectic factors could be transferred between tumor-bearing rats to non-tumor-bearing rats when both are connected surgically via circulation, indicating the humoral nature of these circulatory factors. Succeeding investigations showed that humoral factors are secreted either directly by cancer cells or by stromal cells in the tumor microenvironment, or by distant organs. Primary tumors, as well as metastatic cachexia models, have revealed that cachexia-induced circulating components include hormones, growth factors, metal ions, and both pro- and anti-inflammatory cytokines.84,85 Third, these circulating factors cause cachexia by two separate pathways, either directly by interacting with myocytes and activating muscle catabolic pathways and by restricting muscle protein synthesis, or indirectly through metabolic changes in secondary organs, which leads to muscle atrophy.84–86 Table 2 summarizes the mechanism of cachexia.13,14,34,87–124 The broad picture of CC is summarized in Figure 1.
Cancer cachexia models and mechanisms.

FATP1: fatty acid transport protein 1; CD36: cluster determinant 36; UCP1: uncoupling protein 1; p-p38: phospho p38; PPARγ: peroxisome proliferator-activated receptor gamma; p-PPARγ: phospho peroxisome proliferator-activated receptor gamma; UCP3: uncoupling protein 3; ERK: extracellular signal-regulated kinase; p-ERK: phospho extracellular signal-regulated kinase; P4HAI: prolyl 4-hydroxylase subunit alpha 1; PKM2: pyruvate kinase M2; ARHGDIB: rho GDP dissociation inhibitor beta; PGK1: phosphoglycerate kinase 1; LDHA: lactate dehydrogenase A; ALDA: aldolase, fructose bisphosphate A; GPD2: glycerol-3-phosphate dehydrogenase 2; ENO1: enolase 1; TPI1: triosephosphate isomerase 1; PGAM1: phosphoglycerol mutase 1; EEF1D: eukaryotic translation elongation factor 1 delta; PRDX1: peroxiredoxin 1; CAT: catalase; SPARC: secreted protein acidic and cysteine rich; COL1A1: collagen type 1 alpha 1 chain; COL1A2: collagen type 1 alpha 2 chain; Cav-1: caveolin 1; PYC: pyruvate carboxylase; G6PD: glucose-6-phosphate dehydrogenase; 4-HNE: 4-hydroxynonenal; ATP: adenosine triphosphate; SDS-MYL1: SDS-soluble myosin light chain 1; IL-6: interleukin-6; MURF1/TRIM63: muscle-specific RING finger protein 1/tripartite motif containing 63; LIF: leukemia inhibitory factor; STAT3: signal transducer and activator of transcription 3; p-STAT3: phospho signal transducer and activator of transcription; p-AMPK: phospho AMP-activated catalytic subunit; mTOR: mechanistic target of rapamycin kinase; p-mTOR: phospho mechanistic target of rapamycin kinase; Pax7: paired box 7; LC3bII/LC3BI: light chain 3 B 1/light chain 3 B 2; PBE: peroxisomal bifunctional enzyme; CPT1α: carnitine palmitoyltransferase 1 alpha; BAT: brown fat tissue; ZAG: zinc-alpha-2 glycoprotein; FDG: fluorodeoxyglucose; LDL: low-density lipoprotein; Ddit4/REDD1: DNA damage inducible transcript 4/regulated in development and DNA damage responses 1; Akt1: Akt serine/threonine kinase 1; p-Akt1: phospho Akt serine/threonine kinase 1; p-4E-BP1: phospho eukaryotic translation initiator factor 4E binding protein 1; p-p70S6K: P70 ribosomal S6 kinase; PDPK1: 3-phophoinositide dependent protein kinase 1; H2O2: hydrogen peroxide; MFN2: mitofusin 2; DRP1: dynamin-related protein 1; FIS1: fission mitochondrial 1; PINK: PTEN induced kinase; PRDM6: PR/SET domain 6; Bcl-2: B-cell CLL/lymphoma 2; TGF-β: transforming growth factor–beta; MHC: myocin heavy chain; MyoD1: myogenic differentiation 1; MAPK: mitogen activated protein kinase; TNF-α: tumor necrosis factor–alpha; ROS: reactive oxygen species; GSH: glutathione; gp130: glycoprotein 130; cavin-3: caveolae associated protein 3; EGFR1: epidermal growth factor receptor 1; HIF1-α: hypoxia inducible factor subunit alpha; pS6K: phospho S6 kinase; PTEN: phosphatase d tensin homolog; HSL: hormone sensitive lipase; p-HSL: phospho hormone sensitive lipase; PTHR: parathyroid hormone receptor; PTHrP: parathyroid hormone receptor–related protein; BM-MNCs: bone marrow–derived mononuclear cells; NFATc1: nuclear factor of activated T cells 1; RAGE: receptor for advanced glycation end product; S100B: S100 calcium binding protein B; HMGB1: high mobility group box 1; IL-3: interleukin-3; IL-9: interleukin-9; IL-12p40: interleukin-12 subunit p40; IL-17A: interleukin-17A; IFNγ: interferon gamma; IL-1α: interleukin-1 alpha; IL-1β: interleukin-1 beta; SAA: serum amyloid A1; PON1: paraoxonase 1; PCB: pyruvate carboxylase; G6pase: glucose 6 phosphatase; ATGL: adipose triglyceride lipase; IGF1R: insulin like growth factor 1 receptor; IR: insulin receptor; GLP1: glucagon like peptide 1; Apo CII: apolipoprotein C2; Apo CIII: apolipoprotein C3; PPAR-α: peroxisome proliferator-activated receptor alpha; ACADM: acyl CoA dehydrogenase medium chain; HMGCS2: 3-hydroxy-3-methylglutaryl-CoA synthase 2; LCN2: lipocalin 2; MAFBX/FBXO32: muscle atrophy F-box protein/F-box protein 32; FOXO1: Forkhead box O1; BNIP3: Bcl-2 interacting protein 3; CTSL: cathepsin L; GABARAPL: GABA type A receptor associated protein like; Sirt1: sirtuin 1, NOX4: NADPH oxidase 4; ZIP4: Zrt/Irt-like protein 4; cAMP: cyclic adenosine mono phosphate; E214K: ubiquitin conjugating enzyme E2 14 kDa; USP2: ubiquitin specific peptidase 2; UBC: ubiquitin C; Pax3: paired box 3; MYH2: myosin heavy chain 2; TNNC1: troponin C1; TPM3: tropomyosin 3; TCAP: titin-cap; TNF-20: tumor necrosis factor–20; IL-15: interleukin-15; PLA2G7: phospholipase A2 group 7.
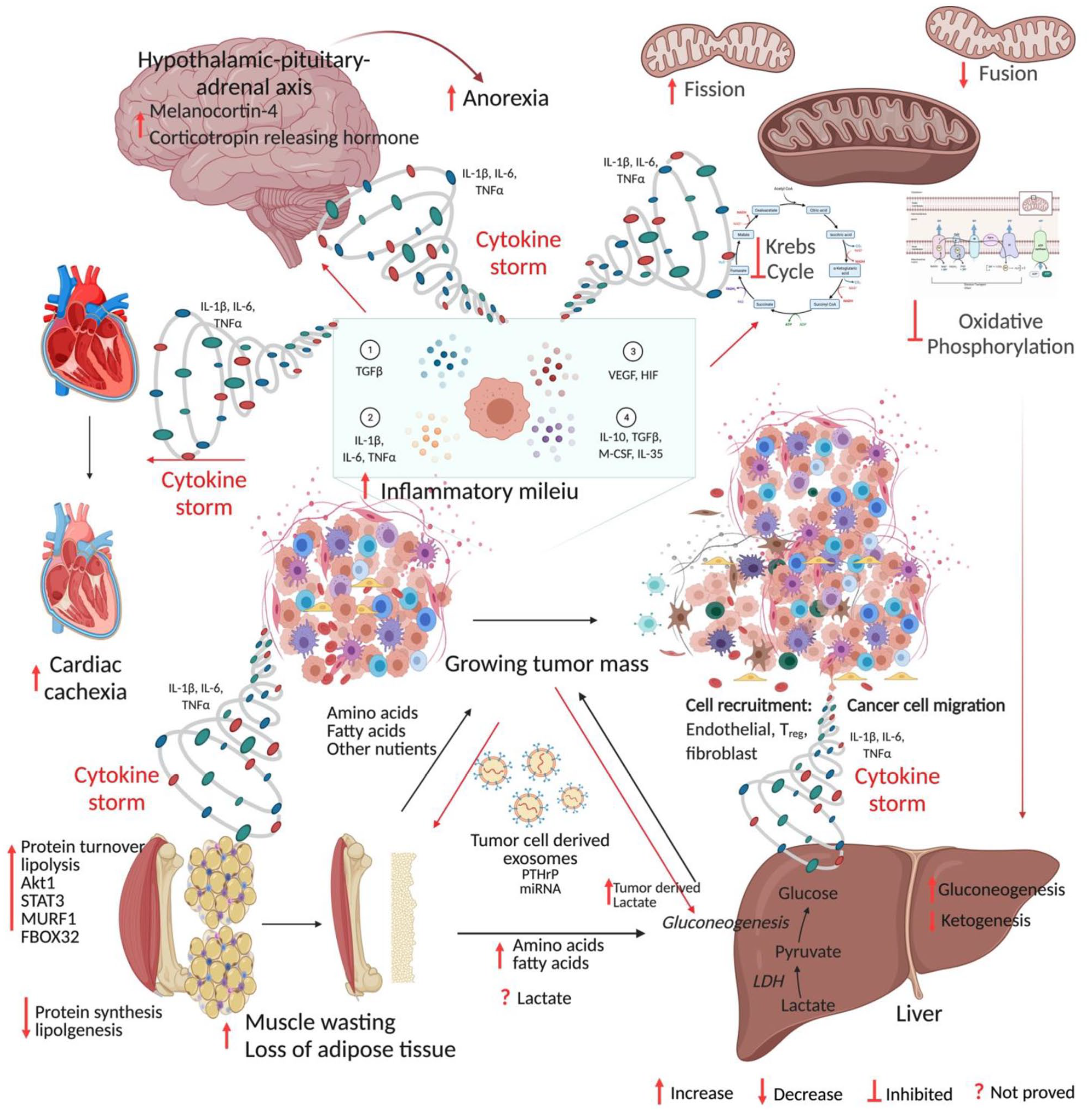
Mechanism of cancer cachexia. Anabolic tumor tissue continuously demands glucose, amino acids, fatty acids, and other nutrients. The cytokine storm generated from the growing mass of tumors acts on host tissue leading to catabolism and cachexia. The figure was created in BioRender.com. (A color version of this figure is available in the online journal.)
Systemic metabolic dysfunction
Cancer cells remodel their metabolic processes to meet the increased demand of proliferative and bioenergetic needs while disrupting host systemic metabolism.1,85 As mentioned above, protein homeostasis is skewed toward the elevated breakdown of muscle proteins and reduced synthesis. 35 Constitutively active ubiquitin-proteasome and insulin signaling pathways are primarily responsible for this. Under anabolic conditions, the PI3K–Akt pathway protects the muscle from undergoing atrophy by inhibiting Forkhead box O (FOXO)-mediated activation of the genes encoding MURF1 and MAFBX or FBXO32. Akt also stimulates the serine/threonine-protein kinase mTOR complex 1 (mTORC1), which activates the S6 kinase-1 (S6K1), leading to anabolic effects on muscle tissues.35–37 Reduced Akt activity has been reported in CC patients to cause the FOXO proteins to be dephosphorylated, allowing transcription of Tripartite motif containing 63 (TRIM63) and FBXO32, which in turn is involved in the myosin heavy chain degradation.36,37 During the progression of CC, hyperactivation of genes encoding MURF1, MAFBX, FBXO30, FBXO31, and increased turnover of myofibrillar proteins were reported, which results in muscle wasting.125,126 Interestingly, it was shown that CC patients often present insulin resistance.38,39 Besides controlling the carbohydrate metabolism, insulin modulates muscle protein breakdown and synthesis to maintain stable blood glucose levels. Physiologically, insulin resistance speeds up muscle proteolysis by suppressing the PI3K–Akt pathway and counter activation of ubiquitin-proteasome pathway.40,41
This mechanism is important in CC since insulin resistance and/or disrupted insulin signaling has been shown in CC murine and Drosophila models in vivo.127–130 Furthermore, in the murine colon 26 (C26) CC model, treatment with the insulin sensitizer rosiglitazone reduced weight loss and anorexia, and in the hepatoma rat cachexia model, this drug hampered weight loss and prolonged survival.131,132 In the Walker 256 cancer, a commonly used in vitro cachexia model, insulin resistance and muscular breakdown were also exacerbated by impaired insulin synthesis from the pancreas. 130 Recent studies involving the fruit fly Drosophila melanogaster conducted by Figueroa-Clarevega and Bilder 129 revealed that tumors release ImpL2, an insulin-like binding protein and a powerful antagonist of insulin signaling, causing systemic metabolic disruption and muscle atrophy. Besides ubiquitin and insulin signaling, a growing body of evidence suggests that the upregulation of autophagy pathways is also crucial for muscle wasting.56,71 A study conducted on lung cancer cohorts presented elevated autophagy mediators such as Bcl2-interacting protein 3 (BNIP3), LC3B (light chain 3 beta), and transcription factors that promote autophagy such as FOXO1. 133 Tardif et al. 134 reported autophagy pathway deregulation in cachectic esophageal cancer patients versus non-cancerous subjects. In addition, Johns et al. 135 documented that increased expression of autophagy-related genes such as microtubule-associated proteins 1A/1B light chain 3B (MAP1LC3B), autophagy protein 5, and beclin 1 in muscles was associated with the CC. Finally, Stephens et al. 136 showed elevated expression of GABA type A receptor associated protein like 1 (GABARAPL1), an autophagy inducer, in cachectic gastrointestinal cancer patients (n = 92) compared to healthy controls. Taken together, these studies indicate that derailed insulin signaling in cancer can have a deleterious influence on muscle hypertrophy and function.
Inflammatory mediators
As cancer advances, cancer cells, infiltrated immune cells, and other stromal cells in the tumor microenvironment secrete enormous amounts of cytokines into the circulation. 137 A vast body of research has established that cytokines including TNF-α, transforming growth factor–β (TGF-β), and IL-6 stimulate muscle fiber disintegration. TNF-α and TNF-related weak inducer of apoptosis (TWEAK) have been shown to directly activate the NF-κB pathway in differentiated muscle cells resulting in the activation of E3 ligases and proteasome-mediated muscle catabolism.138,139 Guttridge et al. 65 showed that NF-κB-induced suppression of muscle cell terminal differentiation is through loss of MyoD. In addition, Fukawa et al. 102 demonstrated that exposure of muscle cells to cancer cell-conditioned medium containing inflammatory molecules, including TNF-α, IL-1β, IL-6, IL-8, LIF and angiogenic factor VEGF, induced fatty acid oxidation resulting in oxidative stress via activating the p38 stress response pathway and impeding the growth of myotube. Tumor-derived IL-6 has been shown to reduce ketogenesis by suppressing peroxisome proliferator-activated receptor–alpha (PPAR-α) in vivo resulting in the marked elevation of glucocorticoids, systemic metabolic stress, reduced food intake, and poor response to anti-cancer immunotherapy. 114 Furthermore, chronic IL-6 is sufficient to induce cachexia in mice, lipolysis in cultured adipocytes, and atrophy in myocytes.12,14,140 Rupert et al. 116 demonstrated that depletion of IL-6 from the malignant pancreatic cells resulted in a dramatic reduction in adipose tissue wasting, myosteatosis, dysregulated metabolism, and eliminated muscle atrophy. Pharmacological inhibition of STAT3 activation has been shown to suppress caspase 3 and ubiquitin-proteasome system resulting in prevention of muscle protein breakdown and cachexia. 66 Recently, Niu et al. 141 demonstrated that HSP90-mediated STAT3 activation induces muscle wasting, weight loss, and catabolism in both in vitro and in vivo models of colon adenocarcinoma, and pharmacological inhibition of HSP90 reversed this effect. Similarly, cancer cachexia has been linked to multiple members of the TGF family. Myostatin (MSTN), for example, interacts with the activin type II receptors ACVR2 and ACVR2B, and stimulates the SMAD2/3 signaling pathway in normal mice, causing significant fat and muscle loss. In several mouse tumor models, administration of ACVR2B prevented cancer cachexia.142–144 Metastatic lesions have been shown to induce the secretion of TGF-β from the bone matrix into circulation, which then stimulates SMAD2/3 in skeletal muscles, in turn inducing the transcription of NADPH oxidase 4 (Nox4). 145 Nox4 induces oxidation and stabilization of ryanodine receptor 1 (Ryr1) leading to aberrant calcium leakage from muscle endoplasmic reticulum, subsequently leading to muscle weakness. 145 Two more TGF superfamily members, namely, growth differentiation factor 11 (GDF11) and GDF15, have subsequently been established as mediators of appetite and cachexia indirectly by controlling the food intake through their action on the hypothalamus.146–148 Elevated serum level of GDF15 was shown to be correlated with increased incidence of CC and poor patient outcome in pancreatic cancer. 63 In addition, transcription factors such as NF-κB, STAT3, and CAAT/enhancer-binding protein-β regulate expression of E3 ubiquitin ligases and autophagy genes.55,65,66,139,149 These studies suggest that inflammatory mediators possibly cause skeletal muscle weakness and atrophy to accelerate CC-associated muscle dysfunction.
Extracellular vesicle mediated regulation
Extracellular vesicles (EVs) are secreted vesicles that aid in intercellular communication by carrying DNA, RNA, proteins, lipids, and metabolites. 150 Several experiments have shown that conditioned medium from cancer cell lines, including LLC, H1299, C26, and AGS secreted EVs carrying heat shock proteins such as HSP70 and HSP90, resulting in induction of cachexia symptoms both in vitro and in vivo.151–154 Mechanistic studies conducted by Zhang et al. 155 showed that EV-derived HSP70 and HSP90 activate Toll-like receptor 4 (TLR4) and p38–MAPK pathways resulting in CC, and that this is prevented by either neutralizing or silencing HSP70 and HSP90 in LLC cells. Recently, Hu et al. 106 demonstrated that LLC cell-derived EVs induce lipolysis both in vitro and in vivo by delivering the parathyroid hormone–related protein (PTHrP) which interacts with its receptor parathyroid hormone receptor (PTHR) to exert downstream effects. Similar observations were made by Yang et al. 156 in 2019 in pancreatic tumor xenograft models. This study further showed that a zinc finger transporter ZIP4 stimulated EVs secretion by cancer cells via Ras-related protein 27B (RAB27B) GTPase, and tumor xenograft models bearing ZIP4 knockdown pancreatic cancer cell lines displayed better body weight and survival than mice with functional ZIP4 bearing tumors. 156 Using human pancreatic cachexia inducing cancer cell lines and patient sera, it has been demonstrated that TLR7/8/9 antagonist IMO-8503 reverses tumor-derived EVs-induced myoblast apoptosis and CC. 157 Waning et al. 145 demonstrated that the TGF-β secretion was induced by fusing PDAC-derived exosomes with liver Kupffer cells, which induce cachexia in metastatic bone disease. Tumor-secreted EVs carrying miR-21 stimulated apoptosis in myoblast cells via TLR7 signaling, leading to muscle atrophy. 158 In addition, Miao et al. 101 demonstrated that exosome-derived miR-195a-5p and miR-125b-1-3p downregulated Bcl-2, and thereby induced muscle fiber breakdown and cachexia in a mouse colorectal cancer model. Recently, Gao et al. 159 found that esophageal squamous cell carcinoma–derived EVs carrying prolyl 4-hydroxylase beta (P4HB) induced skeletal muscle cell apoptosis in a mouse xenograft model. Mechanistic studies by the authors have revealed that P4HB induced apoptosis via activating the ubiquitin-proteasomal pathway and regulating the stability of Bcl-2 and phosphoglycerate dehydrogenase. The authors further proved that the inclusion of CCF642, a P4HB inhibitor, suppressed apoptosis of muscle cells in vitro and prevented muscle atrophy and weight loss in an esophageal squamous cell carcinoma–induced cachexia mouse model. 159
These studies have delineated how cancer cells harbor host tissue metabolism by secretory molecules and cargo packaged EVs and skew the entire host metabolism toward catabolism while themselves being anabolic.
Loss of white adipose tissue
Turnover of adipose tissue, especially white adipose tissue (WAT), has been commonly observed in cancer patients. Early research found that knocking down the gene encoding adipose triglyceride lipase (ATGL), an enzyme that catalyzes triacylglycerol hydrolysis, prevented tumor-bearing mice from losing their WAT and reduced skeletal muscle atrophy. 160 During precachexia, elevated lipolysis, energy expenditure, and markers of adipose tissue thermogenesis have been reported. In a variety of in vivo tumor models, including colon, lung, liver, and pancreatic cancer models, browning of WAT was discovered to be the primary response during CC development that induces lipid mobilization.21,161 Mechanistic studies have shown that pro-inflammatory cytokine IL-6 induces uncoupling protein 1 (UCP1) in WAT, promoting thermogenesis by uncoupling mitochondrial respiration from adenosine triphosphate (ATP) production. 162 Blockade of inflammation either by blocking IL-6 signaling or by blocking β-adrenergic neurons or by using anti-inflammatory treatments resulted in a remarkable reduction of WAT browning. 21 In the in vivo LLC model, tumor cell-secreted PTHrP binds to receptor PTHR on WAT and promotes WAT browning, while PTHrP neutralization preserved skeletal muscles, preventing CC. 162
Furthermore, these circulatory factors alter liver functions to meet their energy demand. 84 Bioluminescence-based metabolic imaging and subsequent experimental evidence have shown that elevated lactate levels in primary tumor tissues of human cervical cancer were significantly correlated with increased risk of metastasis and poor outcome. 163 Through gluconeogenesis, the liver converts lactate obtained from the blood into glucose, which is subsequently reintroduced into the circulation and used for energy sources by other tissues. This is referred to as the Cori cycle and is an energy-inefficient process.42,164 Despite the lack of experimental evidence, it is believed that the high metabolic demands of the tumor, together with an intensified Cori cycle, cause a 40% increase in energy consumption in individuals with advanced CC. 164 The amino acids released through muscle breakdown have been postulated to be converted to glucose via gluconeogenesis in the liver during glucose scarcity.165–167 Furthermore, cachectic muscle metabolic profiling from mice with lung cancer revealed low amounts of ketone bodies in serum despite the fact that prolonged starvation usually stimulates ketogenesis. Reduced ketogenesis combined with restricted food intake significantly raised glucocorticoid levels, a trend which was seen in various CC models.114,168 In a genetically engineered lung CC mouse model, augmenting ketogenesis with the PPAR agonist fenofibrate prevented muscle atrophy by lowering systemic glucocorticoids. 168 In addition, tumor-derived IL-6 reduced peroxisome proliferator-activated receptor-α (PPAR-α)-controlled ketogenesis in the colon, liver, and pancreatic cancer models, and the consequent increase in glucocorticoid levels suppressed intratumoral immunity during caloric restriction.114,168,169 During CC, malfunction of these general energy-inefficient metabolic activities in the liver might prolong energy shortage which necessitates additional muscle breakdown.
Thus, cancer cachexia is likely to be governed by an imbalance in the homeostasis of anabolic and catabolic processes.
CNS in cachexia
Our understanding of the CNS role in CC pathogenesis largely depends on observations from animal CC models. An increasing body of evidence suggests that the paracrine inflammatory milieu generated from peripheral inflammation during CPC is amplified and modified within the medio-basal hypothalamus, leading to an alteration in the activity of neurons involved in the regulation of appetite and metabolic processes.60,170,171 Inflammatory stimuli including TNF-α and IL-1β have been shown to initiate a feed-forward loop by acting upon receptors on hypothalamic neurons—pro-opiomelanocortin, agouti-related protein neurons—resulting in acute illness response, loss of appetite, weight reduction, and muscle protein degradation. 61 CNS/IL-1β induced hypothalamic-pituitary-adrenal axis activation evokes rapid muscle atrophy and is blocked by adrenalectomy or by the muscle-specific knockout of glucocorticoid receptors. 61 IL-1β has been shown to stimulate melanocortin-4, corticotropin-releasing hormone, adrenocorticotropic hormone, and cortisol, thereby reducing appetite and promoting the catabolic effects.172,173 To date, the clinical studies on CNS-regulated cachexia are limited to investigations on circulatory levels or administration of inflammatory molecules or neuromodulatory peptides such as ghrelin. 174
Cardiac muscle atrophy
Currently, a countable number of studies are available focusing on the effects of CC on vital organs. Cardiac muscle performs a vital role and was assumed to be spared since it cannot be exploited as amino acid or fat repository like skeletal muscles during normal physiology. Although the cardiac muscle atrophy in CC remains to be evaluated, research on animal cachexia models has shown substantial loss of cardiac muscle fibers and functional impairment of cardiac muscles by echocardiography. 4 In vivo studies have proved that mechanisms that contribute to cardiac muscle atrophy are similar to those in skeletal muscles including elevated protein degradation, reduced protein synthesis, inflammation, and autophagy.175–177 Although adequate knowledge of mechanisms is lacking, it has been shown that RC is significantly associated with cardiac arrhythmias and arrest in cancer patients. 175
Others
Recently, “omic” studies have been extended to find the molecular mechanisms, pathways, and promising biomarkers for CC. Integrative transcriptomic analysis of cachectic muscle and extensive bioinformatics approaches by Niu et al. 99 revealed that 371 genes were up-regulated and 422 were downregulated with the enrichment of genes involved in extracellular matrix reorganization, muscle system processes, muscle differentiation, muscle tissue development, JAK-STAT signaling, cytokine-cytokine receptor signaling, HIF-1 signaling, and so on. Using in vitro knockdown models, authors further showed that p38 induced expression of Ddit4 which in turn inhibited phosphorylation of Akt1, mTOR, 4E-BP1, and p70SK6. 99 Deletion of REDD1 in LLC-conditioned C2C12 myoblasts showed that elevated phosphorylation of FOXO3A, Akt1, mTORC1 prevented cachexia. 105 Furthermore, knockout of lipocalin 2 (LCN2) in pancreatic cachexia mice models showed the suppression of food intake by directly acting on CNS upon crossing the blood-brain barrier. 115 Recently, transcriptome profiling of rectus abdominis muscle from pancreatic cancer patients showed 340 differentially expressed genes, including FOXO1, FOXO3, phosphoinositide-3-kinase regulatory subunit 1 (PIK3RI), glutamate-ammonia ligase (GLUL), interleukin-6 receptor (IL-6R), ZIP14, protein phosphatase 1 regulatory subunit 8 (PPP1R8), apoptosis enhancing nuclease (AEN), coiled-coil domain containing 68 (CCDC68), Wnt family member 9A (WNT9A), protein O-mannosyl transferase 2 (POMT2), sestrin 1 (SESN1), ring finger protein 207 (RNF207), and dystonin (DST) were found to be correlated with an increasing grade of weight loss in cancer patients. 178
Diagnosis and treatment
Diagnosis of CC is challenging due to the display of multiple symptoms and accordingly, each patient must be critically evaluated for BMI, nutritional status, and locomotory index prior to the optimization of precision medicine.3,25 Cachexia might be under-recognized in certain cases including epidemic obesity. Even at the initial time of cancer diagnosis, obese patients can have substantial ongoing muscle depletion known as sarcopenic obesity. In addition to obesity, underlying cachexia can be masked by weight gain due to ascitic fluid accumulation or peripheral edema.74,78,179 Hence, the effective early diagnosis of cachexia includes careful monitoring of body composition, fat mass, and muscle mass. The Patient-Generated Subjective Global Assessment (PG-SGA) is an approved and validated method for malnutrition in patients with CC. 180 PG-SGA contains a comprehensive questionnaire that assesses calorie intake, body weight, muscle mass, body fat mass, temperature, functional status of locomotory organs, and whether systemic or local edema is present. 180 The Mini-Nutritional Assessment (MNA) is a rapid nutritional screening tool that evaluates dietary history, nutritional risk factors, body weight, and mid-arm circumference. 181 However, these screening tools do not measure muscle mass or composition. 182 Regularly used parameters to evaluate body composition in cancer patients include anthropometric methods, bioelectrical impedance analysis (BIA), computed tomography (CT), and dual-energy X-ray absorptiometry (DXA). The anthropometric method evaluates body composition by measuring height, weight, skinfolds, and body circumference. It is one of the oldest and simplest methods, exhibiting poor accuracy, and it cannot distinguish between fat content and lean body mass. 183 BIA, on the contrary, can be used to assess the percentage of total body fat, fat-free mass, and to calculate body fluid based on the electrical properties. 184 Nonetheless, BIA is not as accurate as DXA, which predominantly determines appendicular muscle mass. Although DXA is cost-effective and requires a very low level of radiation, it does not distinguish subsets of adipose tissues into visceral, subcutaneous, and intramuscular. 185 Meanwhile, recent advancements in CT scans allow assessment of axial muscle mass. This approach offers a high level of sensitivity and specificity and is a gold standard for evaluating body composition. 186 Magnetic resonance imaging (MRI) has a higher specificity and accuracy, equivalent to that of CT, in measuring the body composition, and does not expose the patients to ionizing radiation, although it is more expensive than CT.187,188 Upper arm grip assessment, psychosocial and physical activity are the few parameters to measure muscle strength and functionality of patients with CC.3,189 Screening for the novel biomarkers of CC is the research hotspot. A variety of inflammatory markers and cytokines are proposed to be biomarkers of CC, including hemoglobin content, ALB content, CRP, leptin (LEP), adiponectin, ghrelin, insulin-like growth factor-1 (IGF-1), IL-1, IL-6, and TNF-α.190,191 Moreover, a few cancer patients might be less susceptible to cachexia development. For instance, cachexia is less likely in patients with a loss of function mutation in protein P selectin (SELP). 192 Recently, using an aptamer-based discovery platform, Narasimhan et al. 193 demonstrated the expression of 71 proteins that correlated with pancreatic cancer cachexia, including known cachexia markers such as LEP, MSTN, and ALB as well as novel cachexia markers such as lymphatic vessel endothelial hyaluronan captor 1 (LYVE1), complement C7 (C7), and coagulation factor 2 (F2). However, these biomarkers are affected by several factors such as age, sex, inflammation, and other underlying diseases. 194 In order to measure the CC systematically, systematic scoring methods have been evolved including the CC scoring system (CASCO), the modified Glasgow prognostic score (mGPS), and the cachexia staging score (CSS). The score corresponds with either non-malignant CPC or CC and RC and effectively predicts the survival of CC patients.194–197 More recently, Anderson et al. demonstrated that CC patients display reduced total lean body mass, stair climb power (SCP), upper body strength, and bioavailable testosterone, and increased REE, cytokines levels, and fatigue compared to the cancer patients without cachexia and weight stable patients without cancer. This study also showed that SCP is a better marker of CC with 78% sensitivity and 77% specificity at a cut-off of 336 watts. 198
Over the years, studies have proven that CC can be differentiated from the underlying other muscle wasting disorders by the mechanical characteristics, and the targeted therapies that prevented CC prolonged the survival and improved the quality of life, while tumor cells continue to propagate.143,199 Multidimensionality of CC demands personalized and systematized treatment strategies. Nevertheless, RC is rebellious to treat, and palliative treatments are often preferred over the other therapy options. The first line of choice to prevent further deterioration during CC is to treat the patients with catabolism inhibitory drugs alongside nutrition supplementation, exercise, and psychological counseling.2,200,201 Table 3 and Figure 2 summarize drugs under various clinical trials and their mechanisms.91,92,100–102,174,199,202–219
Preclinical and clinical trials to treat cachexia.
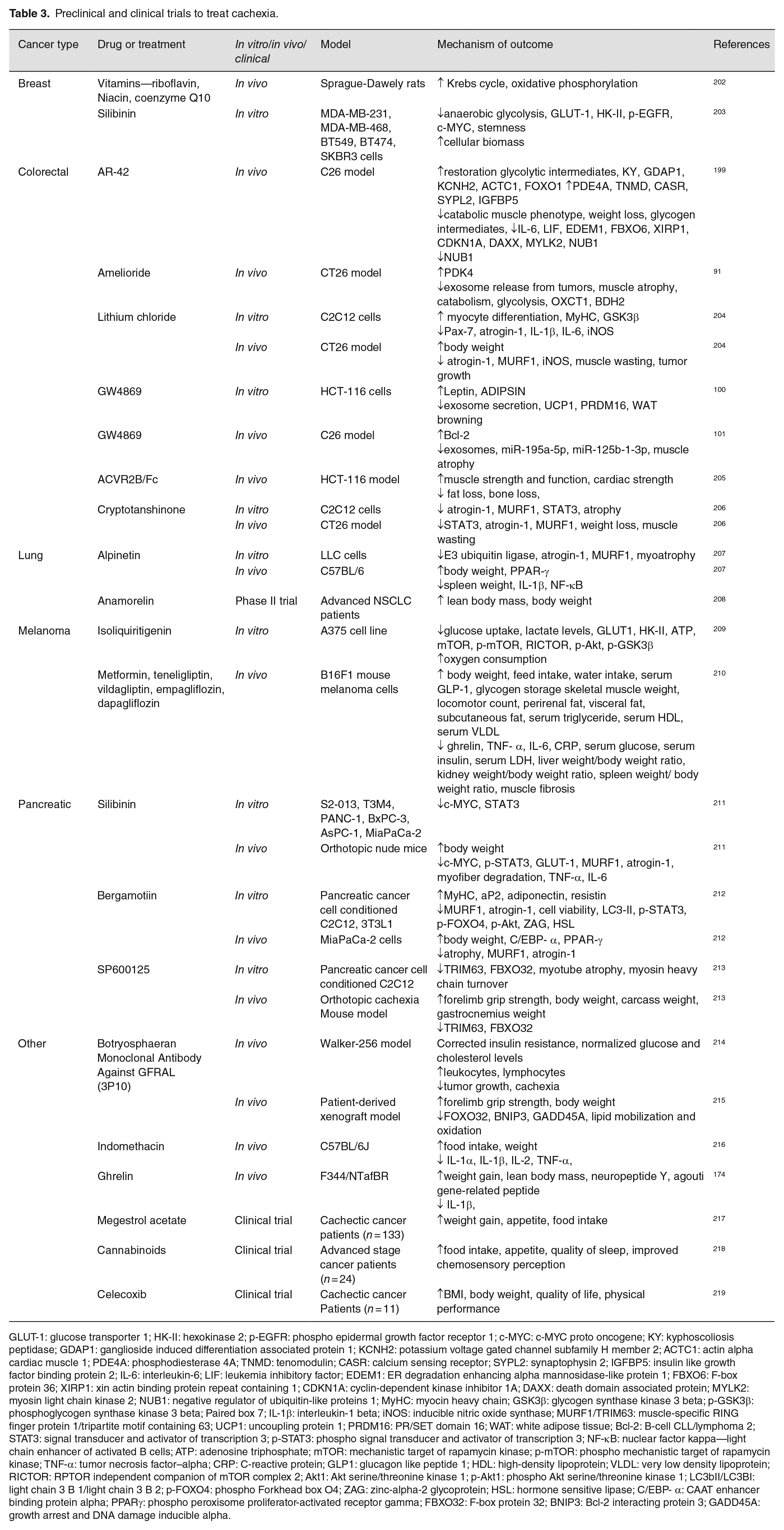
GLUT-1: glucose transporter 1; HK-II: hexokinase 2; p-EGFR: phospho epidermal growth factor receptor 1; c-MYC: c-MYC proto oncogene; KY: kyphoscoliosis peptidase; GDAP1: ganglioside induced differentiation associated protein 1; KCNH2: potassium voltage gated channel subfamily H member 2; ACTC1: actin alpha cardiac muscle 1; PDE4A: phosphodiesterase 4A; TNMD: tenomodulin; CASR: calcium sensing receptor; SYPL2: synaptophysin 2; IGFBP5: insulin like growth factor binding protein 2; IL-6: interleukin-6; LIF: leukemia inhibitory factor; EDEM1: ER degradation enhancing alpha mannosidase-like protein 1; FBXO6: F-box protein 36; XIRP1: xin actin binding protein repeat containing 1; CDKN1A: cyclin-dependent kinase inhibitor 1A; DAXX: death domain associated protein; MYLK2: myosin light chain kinase 2; NUB1: negative regulator of ubiquitin-like proteins 1; MyHC: myocin heavy chain; GSK3β: glycogen synthase kinase 3 beta; p-GSK3β: phosphoglycogen synthase kinase 3 beta; Paired box 7; IL-1β: interleukin-1 beta; iNOS: inducible nitric oxide synthase; MURF1/TRIM63: muscle-specific RING finger protein 1/tripartite motif containing 63; UCP1: uncoupling protein 1; PRDM16: PR/SET domain 16; WAT: white adipose tissue; Bcl-2: B-cell CLL/lymphoma 2; STAT3: signal transducer and activator of transcription 3; p-STAT3: phospho signal transducer and activator of transcription 3; NF-κB: nuclear factor kappa—light chain enhancer of activated B cells; ATP: adenosine triphosphate; mTOR: mechanistic target of rapamycin kinase; p-mTOR: phospho mechanistic target of rapamycin kinase; TNF-α: tumor necrosis factor–alpha; CRP: C-reactive protein; GLP1: glucagon like peptide 1; HDL: high-density lipoprotein; VLDL: very low density lipoprotein; RICTOR: RPTOR independent companion of mTOR complex 2; Akt1: Akt serine/threonine kinase 1; p-Akt1: phospho Akt serine/threonine kinase 1; LC3bII/LC3BI: light chain 3 B 1/light chain 3 B 2; p-FOXO4: phospho Forkhead box O4; ZAG: zinc-alpha-2 glycoprotein; HSL: hormone sensitive lipase; C/EBP- α: CAAT enhancer binding protein alpha; PPARγ: phospho peroxisome proliferator-activated receptor gamma; FBXO32: F-box protein 32; BNIP3: Bcl-2 interacting protein 3; GADD45A: growth arrest and DNA damage inducible alpha.
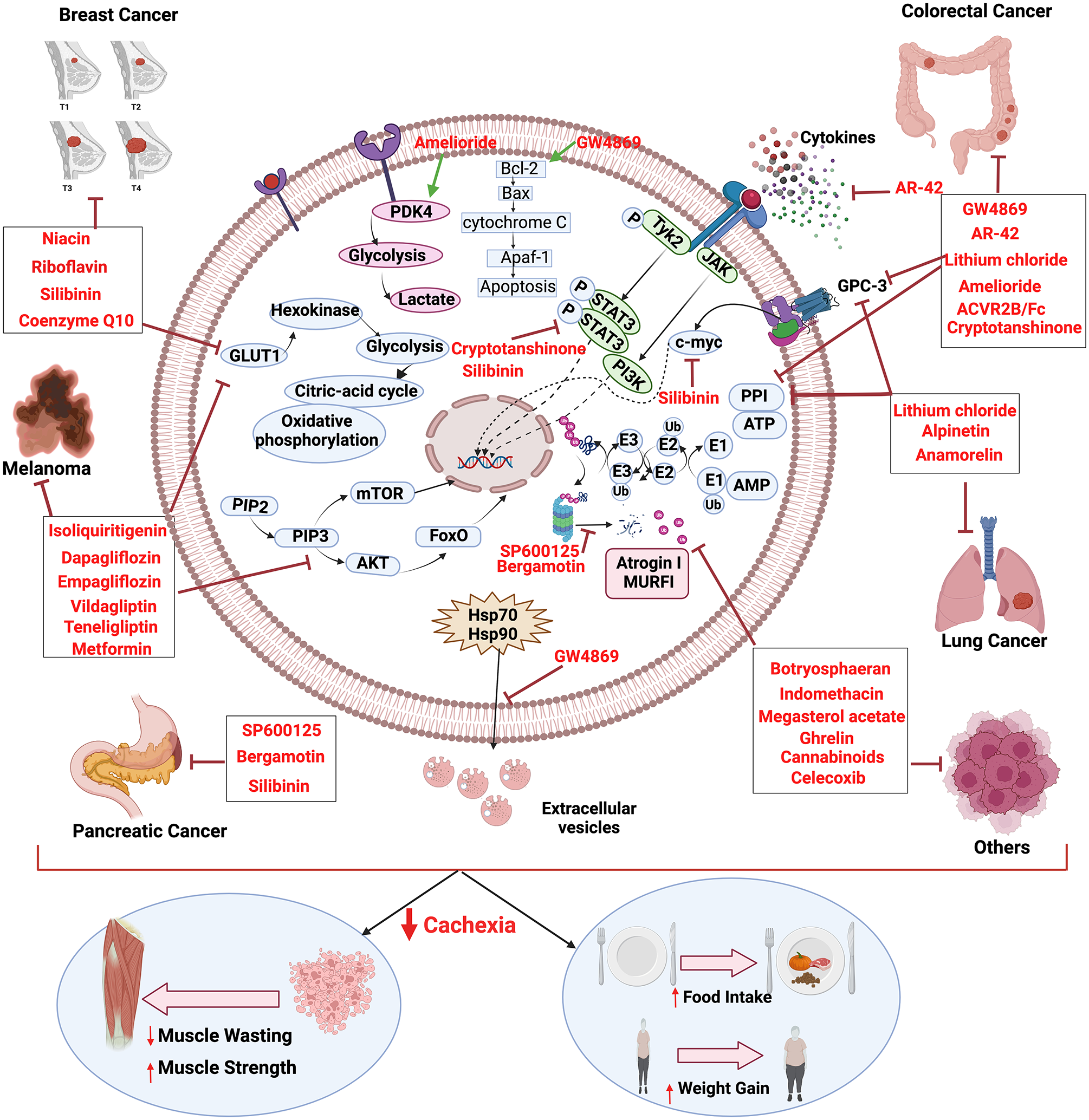
Molecular insight to cachexia and mechanism of cancer cachexia inhibition by various drugs. The figure was created in BioRender.com. (A color version of this figure is available in the online journal.)
ASCO guidelines recommend dietary assessment and psychological counseling as a crucial part of the treatment besides the drug-based treatments. 220 Artificial feeding with priority given to the enteral route has been the preferred way to treat terminally ill cancer patients. 221 It is becoming increasingly obvious that cancer cachexia is a multiorgan disease involving a variety of causes, and hence needs combined therapeutic approaches such as nutrient supply, physical activity, and drugs for its management. 222 Nutritional supplementation is inevitable in cancer patients, as food intake is compromised secondary to anorexia, oral mucositis, and vomiting. 223 For effective anabolic resistance, an increase in caloric intake of 300–400 kcal/day and 50% extra protein intake is essential. 224 High calorie and proteinaceous diet along with oral supplementation of β-hydroxy-β-methyl butyrate (HMB), omega-3 fatty acids, L-carnitine, and eicosatetraenoic acid have shown to be beneficial in patients with CPC and CC.225–229 A study conducted on lung cancer patients showed that oral administration of omega-3 fatty acids increases lean body mass and improves the cachectic conditions in these patients. 230 In addition, pharmacological agents such as those that target proinflammatory cytokines, non-steroidal anti-inflammatory drugs (NSAIDs), and cannabinoids have been widely used to treat CC. 224 Anti-TNF-α agents such as etanercept and infliximab, thalidomide, and pentoxifylline showed a modest gain in lean body mass with no noteworthy clinical benefits.15,16,231–235 Clazakizumab humanized anti-IL-6 monoclonal antibody increased the hemoglobin and ALB levels and relieved fatigue in advanced-stage cancer patients. 236 Administration of ghrelin daily for 8 weeks in low dose (0.7µg/kg body weight) or high dose (13µg/kg body weight) subcutaneously improved the appetite and reduced fat loss in gastrointestinal cancer patients. 237 In a phase II randomized clinical trial, intravenous administration of ghrelin increased the food intake and appetite and reduced chemotherapy-induced nausea in esophageal cancer patients. 238 In addition, progesterone derivatives including megestrol and medroxyprogesterone have been shown to inhibit proinflammatory cytokines such as TNF-α, IL-6, and IL-1 thereby reducing cancer-related anorexia and cachexia.239–241 Various corticosteroids such as dexamethasone (3–6 mg/day), methylprednisolone (12 mg/day), and prednisone (15 mg/day) have been shown to increase appetite and weight gain in cancer patients.242,243 However, these hormonal derivatives have shown long-term side effects, including insulin resistance, myopathy, fluid retention, adrenal inefficiency, and sleep disorders.243,244 In addition, administration of NSAIDs such as celecoxib showed a significant increase in body weight, BMI, and quality of life in head and neck and gastrointestinal cancer patients. 219 A phase III clinical trial showed that administration of phytocannabinoid, tetrahydrocannabinol (THC), increases appetite, body fat, body weight, and quality of life. 245 Recent studies have shown that Kampo medicine, a traditional Japanese medicine, improves CC condition.247–249 Kampo formulae including Hochuekkito (HET), Juzentaihoto (JTT), Ninjin’yoeito (NYT), Seishoekkito (SET), Shosaikoto (SSK), and Rikkunshito (RKT) improved appetite, anemia, protein synthesis, ghrelin levels, and reduced protein breakdown, inflammation, and adipose tissue loss in various preclinical and clinical settings of CC.248–259
Recently, in December 2020, Japan approved anamorelin (non-peptide ghrelin analog) for cancer cachexia treatment based on phase II trials conducted in Japanese advanced gastrointestinal and lung cancer patients, creating the hope for the development of novel drugs.208,260,261 In addition, physical exercise has shown beneficial effects, including reduced systemic inflammation, reduced catabolism, and preserving muscle mass in CC patients along with psychological stability.262–264 Recent studies have highlighted the considerations for multimodal treatment protocol owing to the complexity of pathogenesis in CC. A phase II clinical trial conducted by Solheim et al. 265 used oral nutritional supplements, resistance training, and celecoxib for CC in patients with incurable lung and pancreatic cancers. Next, a randomized controlled clinical trial by Uster et al. 266 implementing nutritional supplements with 60 min of exercise twice a week showed increased protein intake, and reduced nausea and vomiting in palliative cancer patients. Since no major side effects were observed during these studies, multimodal treatment approaches might be considered safe and feasible.
Lack of adequate early diagnosis, prognosis, and therapeutic algorithms creates great difficulty in preventing cachexia development and effective treatments during cancer progression.
Conclusions
Overall, progressive cachexia has deleterious and lethal consequences for cancer patients. The widespread failure of upliftment of CC patients by the usage of anti-cancer therapies led to the realization that critical mediators and mechanisms that induce cachexia could be distinct from those that drive primary tumor progression and metastasis. As mentioned earlier, our current knowledge about cachexia is widely accepted from the studies on genetically engineered and orthotopic animal cachexia models which do not recapitulate systemic and molecular changes occurring in CC patients. Moreover, the vast majority of murine models used for studying CC are LLC in C57BL/6, colon-26 in Balb/c, tumor cell injection, and transgenic mice C57BL/6 APC+/min models. Different strains and different cancer types need to be employed to reveal the relevant cachectic mechanisms. Nevertheless, these observations prompt future studies to aim for screening of relevant early diagnostic and prognostic biomarkers and clinical validation of promising therapies that can effectively treat cachexia alongside the tumor. In addition, it is important to distinguish precachexia from anorexia and later-stage cachexia events. Present knowledge gained from animal studies and a few clinical studies are insufficient to comment on potential mediators of precachexia alone and is a subject of future investigations. Clinical trials on RC patients are lacking due to increased dropout, missing data, and avoiding issues of confounding death. Currently, there is no standard therapy that can effectively reverse the CC. Given the complexity of CC, rigorous preclinical studies in the context of etiology, initiation, progression, and therapy followed by robust clinical validation are all warranted for the effective management of CC.
Footnotes
Authors’ Contributions
MH contributed to the conceptualization, drafting of the manuscript, visualization, and overall editing; UD, SG, and AK supplied critical overall manuscript revision and editing, and ABK contributed to the conceptualization, funding, and overall supervision, and supported review development and overall editing.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Authors acknowledge DBT-AIST International Center for Translational and Environmental Research (DIACENTER) awarded to Prof Ajaikumar B. Kunnumakkara (grant number BT/BI/14/042/2017). Mangala Hegde acknowledges Science and Engineering Board (SERB)-National Post Doctoral Fellowship (NPDF) (PDF/2021/004053). Uzini Devi Daimary and Aviral Kumar acknowledge the Prime Minister’s Research Fellowship (PMRF) program, Ministry of Education (MoE), Government of India for providing them the fellowship.