
Abstract
Type 2 diabetes mellitus (T2D) is nowadays a worldwide epidemic and has become a major challenge for health systems around the world. It is a multifactorial disorder, characterized by a chronic state of hyperglycemia caused by defects in the production as well as in the peripheral action of insulin. This minireview highlights the experimental and clinical evidence that supports the novel idea that intercellular junctions (IJs)-mediated cell–cell contacts play a role in the pathogenesis of T2D. It focuses on IJs repercussion for endocrine pancreas, intestinal barrier, and kidney dysfunctions that contribute to the onset and evolution of this metabolic disorder.
Keywords
Impact Statement
Type 2 diabetes mellitus (T2D) is the most prevalent metabolic disease worldwide associated with obesity. T2D pathogenesis is complex and not fully understood. Experimental and clinical evidence has recently pointed out a role of cell–cell contacts mediated by intercellular junctions (IJs) in the dysfunction of organs (i.e. endocrine pancreas, intestine, and kidney) involved in the onset, evolution, and complication of T2D. IJs, represented by tight, adherens, and gap junctions, would participate in the compensatory hyperplasia process and the secretory dysfunction of pancreatic beta cells at early and late stages of T2D, as well as in the impairment of the intestinal epithelial barrier leading to worsening of the peripheral insulin resistance and in disruption of glomerular/tubular barriers associated with the diabetic nephropathy, one of the complications of T2D. This knowledge opens the possibility that these junctional proteins can be used as target molecules in the gene, cell, and pharmacological therapy of T2D.
Introduction
Cells within a tissue or organ are interconnected with other cells and with the extracellular matrix by specialized membrane-associated structures known as intercellular junctions (IJs) and cell–matrix junctions, respectively. Each different cell type (except the blood cells and spermatocytes) is expected to possess one or more of these junctions. Structurally, IJs are categorized into four distinct components: zonula occludens, or tight junction (TJ), zonula adherens, or adherens junction (AJ), desmosomes, and gap junction (GJ).1–5 TJ is considered the main occluding junction that “seals” cells together, mainly in epithelia, to control the diffusional paracellular transport between the compartments delimitated by this cell sheet. AJ and desmosomes are the main types of anchoring junctions that mechanically attach cells (and their cytoskeleton) to their neighbors. Both TJ and AJ are also involved in determining cell polarity by establishing plasma membrane domains with distinct biochemical composition and function. Cell–cell communication has been assigned to the GJ, which mediates the passage of chemical and electrical signals from one interacting cell to its partners.
In the last decades, the intensive investigation of the molecular biology of IJs has led to the identification of several protein components and an understanding of the mechanisms regulating their organization and function. The molecular structure of these junctions consists of a protein complex composed of integral proteins belonging to different families of cell adhesion molecules that, in the case of TJ, AJ, and desmosomes, can interact with the cytoskeleton through anchoring/adaptor proteins.1,3,4,6,7 Figure 1 shows the main protein interactions at IJs. The current view is that these junctions and their constitutive proteins, besides giving the structural feature necessary for tissue homeostasis, are involved in several signaling processes resulting in crucial events such as gene expression, cell motility, proliferation, differentiation, survival, and death1,3,5,8–10 (Figure 2).
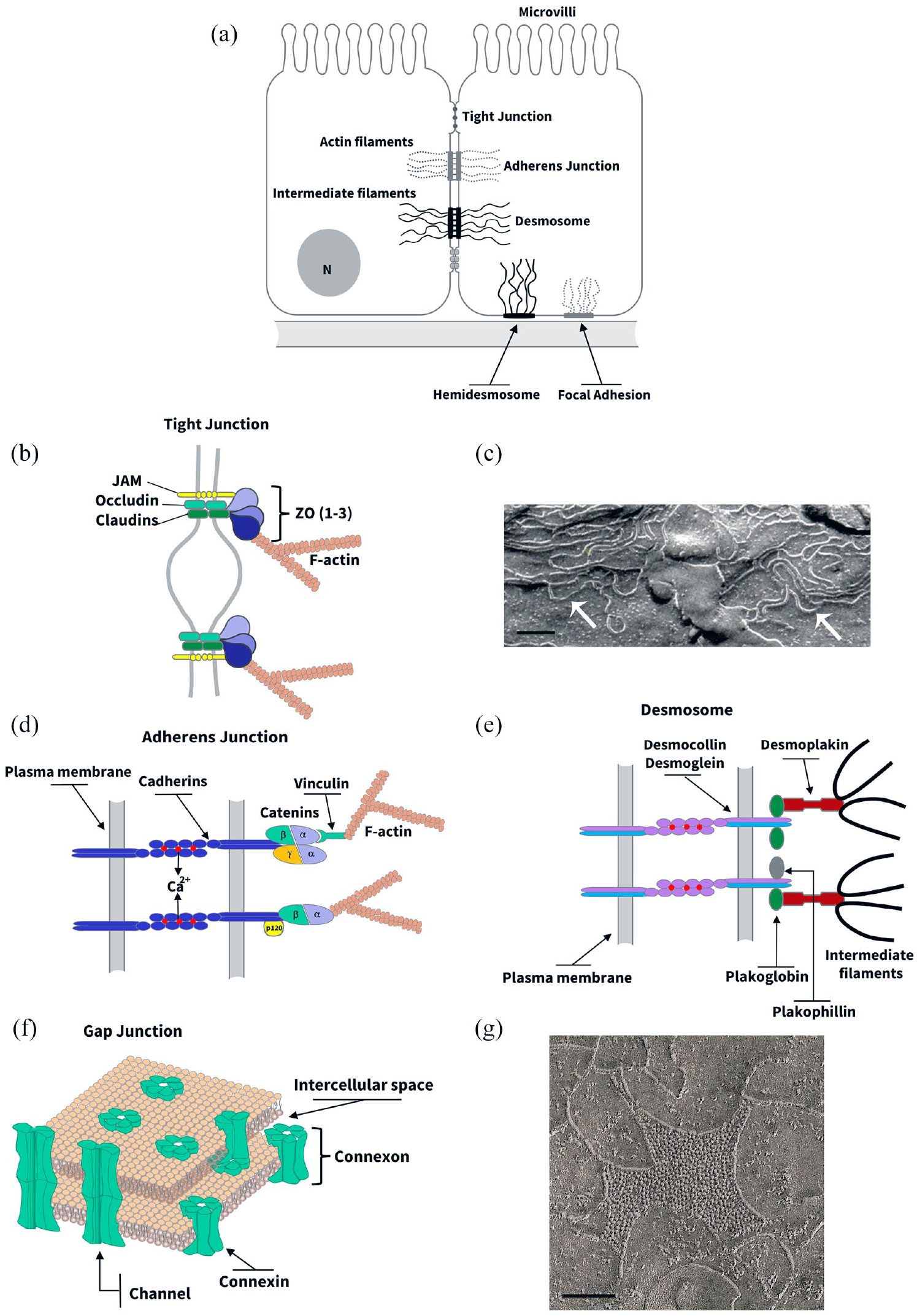
Structure and biochemistry of intercellular junctions. (a) Cell junctions in a hypothetical epithelial cell. (b, d, e, and f) Schematic representations of the molecular structure of tight junction (b), adherens junction (d), desmosome (e), and gap junction (f). (b) Tight junction often appears in transmission electron microscopy as a series of discrete sites of apparent fusion (“kisses”), involving the outer leaflet of the plasma membrane of adjacent cells. At these sites, claudins and other proteins such as JAMs (junction adhesion molecules) and occludin are found. These integral proteins associate with a protein complex at the cytoplasmic side of tight junction that includes ZO-1, ZO-2, and ZO-3, which in turn link them to cytoskeletal F-actin-made microfilaments. (c) The integral TJ proteins interact within the lateral membrane to form anatomizing strands that can be observed in freeze-fracture replicas (arrows). Scale bar, 1 µm. (d) Adherens junction (AJ) appears like a specialized membrane region in which two adjacent plasma membranes is parallel and separated by an interspace 15 to 25 nm wide and filled with a homogeneous, apparently amorphous material of fine density. On the cytoplasmic side of the AJ-associated membrane, there is a thick meshwork of filaments corresponding to the microfilaments of the cytoskeleton. The functions of cell–cell recognition and adhesion attributed to the JA are the result of homophilic interactions between dimers of Ca2+-dependent adhesion molecules belonging to the cadherin superfamily. The cytoplasmic domain is highly conserved among the classical cadherin subtypes and binds directly to several cytoplasmic proteins including β-catenin, γ-catenin, and p120. The β- and γ-catenin interact indirectly with actin microfilaments through α-catenin. (e) Desmosomes are recognized as two straight relatively thick plaques of dense material at two adjacent membranes running parallel to each other and delimitating an intercellular space of 25 nm with filamentous material. A dense fibrillar material, corresponding to intermediate filaments of the cytoskeleton, concentrates at the cytoplasmic sites of the desmosome. Two subtypes of cadherins, desmocollin, and desmoglein are found to mediate desmosomal Ca2+-dependent intercellular adhesion. A cytoplasmic protein complex, containing plakoglobin (i.e. identical to γ-catenin), plakophillin, and desmoplakin among others, mediate the interaction between desmosomal cadherins and cytoskeletal intermediate filaments. (f) Gap junctions (GJ), in transmission electron micrographs of ultrathin tissue sections, appear like regions where the plasma membranes of adjacent cells closely approach each other but are separated by a small gap of 2 to 3 nm. At the molecular level, GJ constitutes a set of intercellular channels, made of integral proteins called connexins, that interconnect the cytoplasm of neighbor cells. (g) Electron micrographs of freeze-fracture replicas of vertebrate junctions show 8.5 to 9.5 nm particles on the P face either free or in plaque-like arrays that correspond to these channels. Scale bar, 0.3 µm. (A color version of this figure is available in the online journal.)
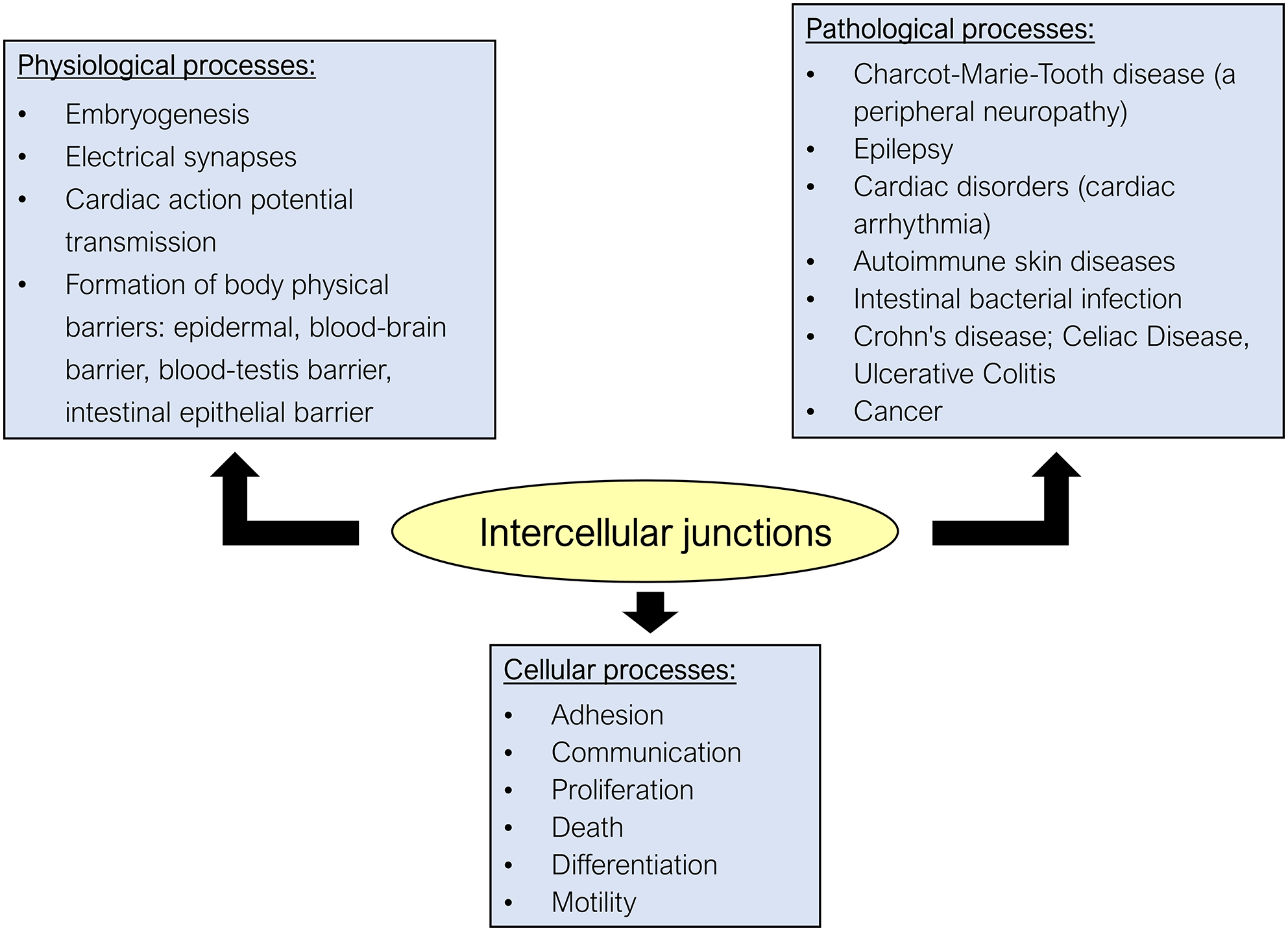
Pathophysiological role of intercellular junctions (IJs). IJs have been considered central components involved in various cellular and physiological processes and their dysfunction is related to various hereditary or nonhereditary diseases. As for cellular processes, in addition to the well-known functions of adhesion and allowing intercellular communication with the exchange of molecules and ions, IJs, through their constitutive proteins, are involved in other important cellular processes. It has been shown that the junctional proteins can migrate to the cytoplasm and nucleus, inducing gene expression of proteins related to cell proliferation, death, and differentiation, and because these junctions are physically associated with the cytoskeleton, they also regulate cell motility. As for physiological processes, the IJs are involved in (1) embryogenesis, because of the importance of cadherins in adherens junction (AJ) in allowing cell recognition and regulating the expression of genes related to cell differentiation; (2) nervous signal transmission, as gap junctions (GJs) form the structure of electrical synapses; (3) cardiac action potential transmission, as the GJ channels, found at the intercalated disks (that also have AJ and desmosomes to provide intercellular adhesion), allow the ionic coupling necessary for the synchronous contraction of the cardiac muscles; and (4) establishment of the epithelial barriers in our body given in part by the tight junction (TJ). Dysfunction of certain IJs is related to the pathogenesis of several hereditary and nonhereditary diseases. Examples are (1) the neuropathy known as CMTX or Charcot–Marie–Tooth linked to the X chromosome, related to a mutation in the gene encoding Cx32 in Schwann cells, resulting in demyelination of peripheral nerves; (2) epilepsy, related to dysfunction of the GJs that form the electrical synapses in the CNS; (3) hereditary heart disease due to deficiency in the expression of Cx43 and desmocollin expressed by the heart muscle, resulting in arrhythmias; (4) autoimmune dermatoses such as pemphigus, which involve the production of autoantibodies that recognize a subtype of desmoglein found in epidermal keratinocytes; (5) diseases related to impairment of the intestinal barrier involving disruption of the TJ (Crohn’s disease, celiac disease, ulcerative colitis, infection by certain intestinal bacteria); and (6) cancer, whose cancer cells typically show loss of intercellular adhesion and also display increased cell motility, as a consequence of the anomalous expression of several junctional proteins that function as tumor markers. (A color version of this figure is available in the online journal.)
IJs are highly dynamic structures that can be regulated by the cell itself or by endogenous and exogenous stimuli (such as hormones and other signaling molecules, toxins and contaminants, certain microorganisms, etc.).3,7,11–14 The regulation of IJs appears to involve one or more of the following processes: (1) alteration of gene expression and/or intracellular degradation of their structural proteins; (2) post-translational modification of junctional proteins (by phosphorylation/dephosphorylation, palmitoylation, or O-glycosylation, ubiquitination); and (3) alteration of the subcellular location of these proteins, through their transport from the cytoplasmic pool to the junctional pool, or vice versa.3,5,14–16 As they are dynamic structures, studies have shown that dysregulation of the IJs and mutations targeting some junctional proteins are linked to several congenital and noncongenital diseases that affect the skin, gut, nervous, and cardiovascular systems1,3,7,10,14 (Figure 2).
This minireview highlights the experimental and clinical evidence that supports the novel idea that IJs-mediated cell–cell contacts play a role in the pathogenesis of T2D. It focuses on IJs repercussion for endocrine pancreas, intestinal barrier, and kidney dysfunction that contribute to the onset and evolution of this metabolic disorder.
Pathogenesis of T2D at a glance
Diabetes mellitus is increasing in incidence, prevalence, and importance as a chronic metabolic disease with more than 420 million people affected throughout the world. 17 The two most common forms of diabetes are type 1 diabetes (T1D) and T2D. T1D results from the autoimmune destruction of the insulin-producing pancreatic beta cells. Susceptibility to this diabetes form is inherited, but environmental factors, such as diet, stress, and viral infections, have been proposed to play a modifying, and perhaps even a primary role in the development of T1D. 18 Meanwhile, T2D, which accounts for 90% to 95% of the total diagnosed diabetes cases, is a relatively more complex and heterogeneous condition characterized by increased levels of blood glucose due to an impairment in peripheral insulin action associated with beta-cell dysfunction. T2D pathogenesis is not fully understood, but obesity is the main risk factor, in addition to a sedentary lifestyle associated with poor eating habits (with high consumption of food rich in saturated fatty acids, total fats, besides the inadequate consumption of dietary fiber).17–19 Also, genetic and epigenetic factors seem to contribute to T2D susceptibility.18,20
The main phenomenon in T2D is the peripheral insulin resistance, where tissues do not respond adequately to this hormone, particularly the adipose tissue, muscle, and liver, leading to an increase in the concentration of circulating glucose (hyperglycemia) and in the blood concentration of free fatty acids (FFAs) and cholesterol (dyslipidemia). This hyperglycemia is partially compensated by an increased insulin synthesis and secretion by the pancreatic beta cells and/or by an expansion of beta-cell mass within the endocrine pancreas.19,21,22 These compensatory events mainly occur during the initial phase, known as prediabetes. However, the high secretory activity of beta cells, for an extended period, may result in their functional exhaustion and their death by apoptosis. Therefore, in the late phase of T2D, the patient often needs hormone replacement to control glycemic homeostasis. 19 Genetic studies have identified some candidate genes linked to T2D that affect the beta-cell capacity to compensate the hyperglycemia, such as the TCF7L2, which encodes a key transcription factor of the Wnt signaling pathway, and the CDKN2B (cyclin-dependent kinase inhibitor 2B), both involved in cell cycle regulation, and the KCNJ11 gene, which encodes the Kir6.2 subunit of the ATP-sensitive potassium channel (KATP), which plays a crucial role for insulin secretion.23,24
More recently, it has been suggested that the intestine also plays an important role in the pathogenesis of T2D, as evidenced by the observation of increased intestinal permeability and endotoxemia in diabetic patients and animal models.25–27 According to the current hypothesis, the disruption of the intestinal barrier associated with intestinal dysbiosis would result in the entry into the systemic circulation of LPS, bacterial toxins, and other substances that would trigger or worsen the state of peripheral insulin resistance.
The progression of T2D, especially in cases where hyperglycemia and dyslipidemia are not controlled, leads to impairment of different organs and systems, such as the kidneys (diabetic nephropathy), the retina (diabetic retinopathy), the cardiovascular system, and the peripheral nervous system (diabetic neuropathy).17,28 These systemic complications of T2D have serious health impacts and can be the main cause of premature death (mainly due to cardiovascular and/or renal failure) in the world population.
Cell–cell interactions and failure of insulin secretion by beta cells in T2D
Beta cell is one of the endocrine cell types of the pancreatic islets, which constitute the morphofunctional units of the endocrine pancreas (representing 1–2% of total pancreas mass). Within the islet, this insulin-secreting cell connects, homotypically (with other beta cells) or heterotypically (with the other pancreatic endocrine cells), through IJs. Several in vivo and in vitro studies have demonstrated that the cell–cell interactions within the endocrine pancreas are crucial for proper glucose-stimulated insulin secretion at physiological conditions.19,29–41 The GJ channels, formed of connexin 36 (Cx36),19,29 act by transmitting, between beta cells, the electrical pulses and the cytosolic increase in (Ca2+), triggered by the intracellular metabolism of glucose. These changes in (Ca2+) are crucial for the exocytosis process of insulin granules. 30 Intercellular communication via gap junctions allows the amplification of the insulin secretion process in two ways: (1) decreasing and/or correcting the functional heterogeneity of beta cells, which may differ with regarding the biosynthesis and insulin secretory response to secretagogues, allowing different subpopulations of beta cells to have similar functional responses, and (2) synchronizing the pulsatile electrical activity and insulin secretory dynamics of beta cells across the islet, which is important for amplifying the overall size of in vivo plasma insulin oscillation.30–34 Meanwhile, AJ and TJ and their proteins (cadherins/catenins in AJ, and occludin/ZO-1 in TJ) may influence the beta-cell secretory function by affecting processes such as (1) the beta-cell polarity, that determines a distinct basal domain (enriched in insulin granules), where the beta cells contact the islet vasculature,34–36 as well as the apical domain, where can be found the primary cilia that are important for insulin secretion, 37 (2) the organization of beta-cell cortical actin microfilaments that regulate insulin granule exocytosis,35,38,39 and (3) the gene expression and activity of key signaling proteins for secretion and cell proliferation, differentiation, and survival.35,40,41
As commented earlier, the beta cell plays a central role in the pathogenesis of T2D since is responsible for an adaptive response that involves alteration in its secretory activity and relative mass during prediabetes, and its failure and death contribute to glycemic dysregulation and systemic complications at the advanced stages of this disease.19,42–44 During prediabetes, beta-cell compensation involves enhanced insulin biosynthesis (by the induction of insulin gene expression and insulin mRNA translation), increased responsiveness of the nutrient stimulated-secretion coupling (through the enhancement of the mitochondrial activity, the action potential amplitude, and the Ca2+ channel activity), and/or expansion of beta-cell mass (by hypertrophy and hyperplasia).19,21,43,45,46 In contrast, several of these steps, such as insulin biosynthesis, stimulus-secretion coupling of insulin release, and a sustained increase in beta-cell mass to compensate the hyperglycemia are impaired once T2D is established.19,42–44,47 These latter changes are consequences of mitochondrial dysfunction, increased oxidative stress, and endoplasmic reticulum stress, besides a proinflammatory factor-mediated cell dysfunction/death, which are elicited, at least in part, by chronic exposure to lipids/FFAs and hyperglycemia, among other factors such as the peripheral release of adipokines/cytokines and the insulin resistance itself.18,19,42,43
Experimental evidence has now revealed that IJs and their proteins also participate in beta-cell dysfunction in the context of T2D. It has been demonstrated that a deficiency in beta-cell coupling mediated by GJ occurs already in the initial phase of the disease (prediabetes) 48 followed by impairment of the AJ and TJ structure in more advanced stages, with direct repercussions on the insulin secretion process. 49 We reported that the impairment of intercellular coupling is the result of a decrease in the cell level of Cx36 and a consequent reduction of the number of functional GJ channels at the plasma membrane of beta cells of prediabetic mice fed a high-fat diet for a relatively short period. 48 This reduction in GJ-mediated beta cell–beta cell coupling seen in the prediabetic animal would interfere in the oscillation of the intracellular concentration of Ca2+, and consequently impact the insulin secretion response to glucose during the early stage of T2D.48,50 This idea is further supported by in vitro and in vivo studies showing that the loss/reduction of Cx36 in mice (by conditionally knocking out the gene) and in beta-cell lines (using antisense oligonucleotides or palmitate treatment) results in loss of the beta-to-beta cell synchronization of intracellular calcium changes, associated with deleterious disturbances of basal, and/or stimulated insulin secretion.31,32,51,52 Also, recent work has been shown that caloric diet restriction completely recovers the prediabetes-associated declines in Cx36 gap junction coupling, leading to an improvement of beta-cell Ca2+ dynamics and insulin secretion as well as the reestablishment of glycemic homeostasis in mice. 50 The finding that in vitro exposure of beta-cell lines and human islets to high concentrations of glucose and fatty acids or, to low concentrations of inflammatory cytokines down-regulates Cx36 expression via cAMP/PKA- and NOS-dependent mechanisms, suggests a possible role of hyperglycemia, hyperlipidemia, and inflammation in the impairment of GJ communication during prediabetes.53–55
Interestingly, in vivo loss of Cx36 further sensitizes beta cells to cytotoxic conditions, including proapoptotic cytokines which concentrate in the islet environment, particularly at the beginning of T1D and later in T2D. 56 Conversely, mice over-expressing Cx36 appear fully protected against the same insults, suggesting that Cx36 is also relevant for the survival of beta cells, 57 and a decrease in its expression may contribute to the reduction of the beta-cell mass seen at advanced phases of diabetes. 56
Although the essential proof of concept of a link between Cx36-made channels and T2D pathogenesis in humans is still lacking, two findings strengthen this possibility. First, the human Cx36 gene is located on chromosome 15q14, a locus that genome-wide scans linked to diabetic phenotypes in different human populations, indicating the involvement of Cx36 in the chain of events that lead to T2D susceptibility.58–60 Second, glibenclamide, a sulphonylurea, Exendin-4, a GLP-1 receptor agonist, and GLP-1 itself, compounds that have been successfully used to improve glycemic control in patients with T2D, induce upregulation of Cx36 gap junction coupling in rodent and/or human beta cells, as well as protect against palmitate-, and cytokine-induced islet dysfunction.52,56,61,62
Besides the GJ dysfunction, in obese and diabetic animals fed for a long time a high-fat diet, we also observed changes in the structure of AJ, which included the redistribution of associated N- and E-cadherins and α-catenin (from intercellular contact to the cytoplasm of endocrine cells), with an accumulation of submembranous actin microfilaments. 49 We suggest that this remodeling of the actin cortical cytoskeleton in the beta cell, resulting from the decrease in the junctional content of proteins in AJ, created a barrier hindering the access and subsequent the docking of insulin granules to the plasma membrane, which would explain, in part, the significant decrease in the secretory capacity of these cells in diabetic animals. 49 Supporting this idea, several works, mainly in vitro, have indicated a positive relationship between the degree of expression/junctional content of cadherins and catenins and the level of glucose-stimulated insulin secretion in beta cells.35,39,63–65 Parnaud et al. 65 elegantly demonstrated, using chimeric proteins made of E-, or N-cadherin ectodomains fused to the Fc fragment of immunoglobulin (E-cad/Fc or N-cad/Fc) attached to an inert substrate, that specific ligation of cadherins can increase insulin secretion of single beta-cells in response to glucose, which was associated with an asymmetrical redistribution of cortical actin, characterized by its reduction at the cell–cell adhesion site.
We also reported the participation of β-catenin in the beta-cell hyperplasia, compensating for the peripheral insulin resistance during prediabetes, probably in conjunction with other signaling pathways such as the JAK/STAT, PI3K/Akt, and ERK/MAPK pathways.66–68 β-Catenin, in addition to being a constituent protein of the AJ cytoplasmic complex, acts as a transcriptional cofactor of the Wnt/β-catenin signaling pathway. This pathway is known to influence multiple processes, such as cell proliferation and differentiation during animal development, directly participating in the morphogenesis of certain tissues/organs. 69
We demonstrated that hyperplastic pancreatic islets from prediabetic mice show activation of the Wnt/β-catenin pathway, as revealed by the nuclear translocation of the active β-catenin protein, associated with a significant increase in protein content and β-catenin gene expression, as well as of cyclins D1 and D2, c-Myc and insulin 2 (target genes of the canonical Wnt pathway). 22 Furthermore, we identified that pancreatic islets are the source of synthesis of several types of Wnts, being the Wnt3a and Wnt5b islet levels increased in hyperplastic islets from prediabetic mice. 70 Co-cultivation of pancreatic islets, isolated from control and prediabetic mice, and the MIN6 beta-cell line (Mouse Insulinoma clone 6) resulted in a significant enhancement of cell proliferation in MIN6 cells, which was partially inhibited by antagonists of the Wnt pathway. 70 Based on these data, we suggest the novel idea that Wnts secreted by pancreatic endocrine cells would participate, via autocrine or paracrine action, in beta-cell expansion in high-fat-induced prediabetes. Despite the well-known differences in functional characteristics between human and mouse beta cells, inclusive when it comes to beta-cell mass regulation, the participation of the Wnt/β-catenin pathway in human beta-cell proliferation in vitro has also been suggested by others,71,72 although its involvement in the context of type 2 prediabetes is still largely unexplored.
There is also evidence that some of the known genetic risk factors for beta-cell dysfunction in humans may be acting, at least in part, by regulating adherens junctions in beta cells.64,73–76 One example is the study showing that the beta-cell expression of the dominant-negative form of HNF-1α, which causes the human maturity-onset diabetes of the young-3 (MODY3), resulted in the onset of diabetes with impaired glucose-stimulated insulin secretion, associated with a reduced expression of E-cadherin in beta cells of transgenic mice. 64 Another example is the close link established between the degree of gene expression of variants of the transcription factor of Wnt/β-catenin pathway, the TCF7L2, and T2D in human populations of different ethnic descent.73–76 Therefore, the TCF7L2 gene is regarded, so far, as one of the most influential genes in determining the genetic susceptibility for T2D in human beings.
Tight junction-mediated intestinal barrier disruption and T2D
Clinical and experimental studies point out intestinal involvement in the evolution of T2D. One of the first studies on this subject was conducted by Cani et al.,25,26 who demonstrated that the intake of a high-fat diet induces changes in the luminal microbiota associated with an increase in intestinal permeability that facilitates the entry of LPS, increasing plasma levels of this component, and leading to endotoxemia in diabetic mice. Larsen et al. 77 also observed a change in the microbiota composition of T2D patients compared to healthy individuals, which was associated with increased intestinal permeability. This was followed by other clinical works that reported a link between T2D and impaired intestinal barrier.78–81 The current hypothesis, which tries to explain the involvement of the intestinal microbiota in the pathogenesis of the two forms of diabetes (type 1 and type 2), proposes that the change in the composition and functionality of this microbiota, due to, at least in part, a diet rich in lipids and poor in fiber, is associated with impairment of the intestinal barrier.25–27,82–85 This, in turn, would cause the systemic entry of bacteria and their products, in addition to food allergens, resulting in hypersensitivity of the immune system in the case of T1D, or a state of endotoxemia and low-grade systemic inflammation, contributing to peripheral insulin resistance in T2D.25–27,82–85 Therefore, according to this hypothesis, the intestinal barrier would constitute a central factor in the relationship between microbiota and metabolic disturbances in diabetes.
One of the main physical components of the intestinal barrier is the TJ that connects the juxtaposed columnar epithelial cells that form the intestinal epithelium.6,86 Other elements of this barrier include the intestinal cells themselves, the mucus secreted by goblet cells, and the immune system associated with the intestinal mucosa. The TJ constitutes the anatomical site of the paracellular epithelial barrier that limits the diffusion transport of substances through the intercellular spaces between the luminal and internal environments.6,86 Although studies focusing on the intestinal TJ regulation during T2D are scarce, it has been demonstrated that T2D animals display reduced expression of TJ-related genes (i.e. claudins-1, -3, JAM-1, ZO-1, and occludin) in the intestinal mucosa, along with an increased plasma level of inflammatory cytokines and LPS.26,87,88
One important question that has been raised is whether (or not) gut permeability is the cause, the consequence, or a mixture of these two in T2D.89,90 In recent works, we have tried to address the cause-and-effect relationship between TJ-mediated intestinal permeability and the evolution of this metabolic disease, as well as the time course of intestinal barrier dysfunction and the contributions of individual intestinal segments toward the regulation of overall gut permeability. We demonstrated the structural and functional disruption of the intestinal epithelial barrier mediated by TJ (particularly in the proximal intestine), even before the onset of prediabetes and endotoxemia and in the apparent absence of local and systemic inflammation, suggesting a contributing role in triggering/worsening of T2D in mice fed a high-fat diet for different periods (15, 30 and 60 days).91–93 When analyzing the structure of TJ in the intestinal epithelium of high fat-fed prediabetic mice, by immunofluorescence and Western Blot, we observed a cellular redistribution of TJ proteins (claudins-1, -3, occludin, ZO-1) and a significant decrease in their junctional content, indicative of impairment of the intestinal paracellular barrier, which was more evident in the proximal intestine (duodenum and jejunum) than in the distal one (ileum and colon), which concentrates a large part of the intestinal microbiota. This general decrease in junctional content of TJ proteins in intestinal epithelia, seen in prediabetic mice, did not involve changes in gene/protein expression, but may reflect a reorganization of the TJ structure, where part of its protein content was removed from the intercellular contact region, probably as a result of post-translational modification of these proteins.15,16,94–96 This impairment of the intestinal paracellular barrier in prediabetic animals was associated with an increase in intestinal permeability to low molecular weight molecules (~400 Da, i.e. Lucifer Yellow), but not to large molecules (⩾4000 Da, i.e. FITC-Dextran). This increase in intestinal permeability to some molecules occurred before the establishment of endotoxemia (assessed by plasma LPS level) and is accompanied by an apparent absence of local and systemic inflammation (assessed by plasma/intestinal zonulin and TNF-α level quantifications, besides by histological analysis of intestine fragments).92,93
In an in vitro model of the intestinal epithelial barrier, exposure of Caco-2 cell monolayers to intestinal luminal content, particularly that obtained from prediabetic animals, resulted in a significant decrease in transepithelial electrical resistance and increased paracellular permeability to markers, which was associated with a significant decrease in the junctional content of TJ proteins (such as claudins-1, -2 and -3, occludin, and ZO-1) (assessed by immunofluorescence) and reduced protein expression (assessed by Western Blot) of some of these proteins. 97 Corroborating our in vivo studies, the luminal content of the small intestine induced more severe alterations in the structure and function of the paracellular barrier in Caco-2 cell monolayers, when compared to the luminal content isolated from the large intestine. 97 Therefore, our work suggests that the increase in intestinal permeability described in diabetic patients and animal models can be explained by the direct action of components of the modified intestinal lumen on intestinal TJ. Possible candidates of luminal elements that could directly affect the TJ-mediated intestinal epithelial barrier in diabetic individuals are the microbiota-derived products,11,25,26,83 diet-derived compounds, such as high levels of glucose and lipids/fatty acids,98–100 and digestive system-derived secretion, such as proteolytic enzymes of gastric, pancreatic and intestinal origin, 101 and bile acids.88,102
It has been shown that some components that display antidiabetogenic effects also increase the intestinal barrier function. We saw that the treatment with sodium butyrate (a short-chain fatty acid—SCFA) has a beneficial effect both on the claudin-mediated intestinal epithelial barrier and on metabolic/systemic changes found in vivo after high-fat diet feeding. 91 Recent work by Chen et al. 103 demonstrated that the supplementation with the amino acid glycine significantly enhanced the TJ-mediated intestinal barrier and, in parallel, ameliorated the glucose tolerance and systemic inflammation in high-fat diet-fed obese mice. Butyrate is produced mainly from the intestinal microbiota fermentation of undigested carbohydrates/fibers, while glycine is enriched in animal-derived foods but poorly found in plant-sourced foods.104–106 Interestingly, both butyrate and glycine can be found at low levels in plasma/serum of diabetic humans and animals.106,107 In the case of glycine, lower concentrations of this amino acid are negatively associated with insulin resistance and are predictive of T2D. 107 Taken altogether, these findings strengthen the pathophysiological role of intestinal TJ in T2D development/progression and the importance of consumption of a balanced diet, as well as suggest a possible therapeutic use of these diet components in T2D management.91,103,104,106 Interestingly, conventional treatments of the metabolic dysfunctions associated with T2D, such as physical exercises and administration of metformin (an oral antidiabetic drug), have beneficial effects also on butyrate-producing intestinal microbiota, glycine plasma levels, and/or gut TJ barrier function.108–111
Glomerular filtration and tubular barriers in T2D-related nephropathy
Diabetic nephropathy (DN) is one of the main complications of diabetes and can lead to chronic kidney dysfunction. It is estimated that 25–40% of patients with T2D are vulnerable to developing this disease, whose main clinical sign is proteinuria. 112 The pathophysiology of DN is complex and multifactorial, however, hyperglycemia is considered one of the main risk factors for the development of the disease.113,114 This happens because glucose at high concentrations in the bloodstream, for a long time, triggers a series of structural and physiopathological changes in the kidneys, such as disruption of the glomerular filtration barrier (GFB), loss of glomerular glycocalyx, injury of podocytes and renal endothelium, accumulation of extracellular matrix in the mesangium and the tubular/interstitial basement membranes, proliferation, hypertrophy, and/or apoptosis of cells from different segments of the nephron, kidney fibrosis, and activation of inflammatory processes.112,113 Besides hyperglycemia, increased plasma lipid levels (dyslipidemia) seem to be involved in the progression of DN.114,115
There is evidence suggesting that impairment of the renal epithelial barriers is an important factor in the pathogenesis of DN.116,117 These barriers depend on IJs between their cell components, such as the fenestrated endothelial cells, and the podocytes in the GFB at the renal corpuscle, as well as the tubular epithelial cells lining the renal tubules. Podocytes, a highly specialized epithelial cell of mesenchymal origin, are characterized by their foot processes, a network of interdigitating cellular extensions, which support the glomerular capillary loop from the visceral leaflet of Bowman’s capsule and interact at specialized cell-to-cell junctions called slit diaphragms. Slit diaphragms are modified IJ and contain proteins that are found in TJ (i.e. claudins, occludin, ZO-1) as well as in AJ (catenins, P-cadherin) and GJ (Cx43), besides unique membrane/cytoskeletal-associated proteins such as synaptopodin, nephrin, and podocin.116,118,119
In pathologic glomerular proteinuria, such as that seen in DN, there is an increased filtration of macromolecules (typified by albumin and other plasmatic proteins) across the GFB. Decreased expression of claudin-5, ZO-1, podocin, and nephrin, indicative of impairment of GFB, was associated with proteinuria in T2D patients and animal models of T2D (db/db and ob/ob mice), which was reversed by an antiinflammatory compound, mefunidone, the peroxisome proliferator-activated receptor-γ (PPARγ) agonist pioglitazone, and the SGLT2 inhibitor, empagliflozin, that all also have nephroprotective effects on DN.120–125 Downregulation and redistribution of Cx43 expression in podocytes were observed in glomeruli of T2D patients and this alteration was closely associated with the progression of overt diabetic nephropathy. 126 According to the authors, Cx43 redistribution may influence slit diaphragm components through direct interaction with ZO-1 and tubulin in podocyte foot processes, leading to the loss of GFB function. 126
Given the importance of the podocyte slit diaphragm for the GFB structure and proteinuria development, some of their proteins, such as nephrin, podocin, ZO-1, and α-actinin-4, have been even employed as urinary biomarkers of early detection of DN and to differentiate DN phenotypes in patients with T2D.118,127–129
Alterations in the renal tubular epithelial barrier in DN have been also reported, although the studies are scarce and mostly focused on T1D animal models.130,131 One of the only works using a T2D animal model was recently published looking at the claudin expression in glomeruli and proximal tubule, the two major targets for hyperglycemia-induced kidney damage. 125 The authors found that claudin-2 expression in proximal tubules and claudin-5 expression in glomeruli were significantly reduced in both T1D and T2D models, which paralleled with higher proteinuria and impaired sodium and potassium reabsorption, increased hyperglycemia, and higher enzymatic activity of MMP-2 and-9, but lower antioxidant capacity in both models. They hypothesized that these metalloproteinases would promote the proteolytic degradation of claudin-5 and claudin-2 and, consequently, the disruption of the TJ structure, contributing to the increase of glomeruli permeability and decrease of reabsorption of ions in proximal tubule at the early stage of DN. 125 Corroborating this data, a reduction in the expression of claudin-5 and claudin-2/occludin was, respectively, observed in the glomeruli and proximal tubules of T1D animals.130,131 Through immunoprecipitation, it was also demonstrated an increase in claudin-2 nitration, induced by oxidative stress, in these animals. In the distal tubules, there was an increase in the expression of claudin-4 and 8, as well as a redistribution of ZO-1 from the membrane to the cytosol, demonstrated by immunohistochemistry. It was suggested that such TJ-protein alterations are possibly involved in the natriuresis observed during diabetic nephropathy.130,131
In vitro studies relate hyperglycemia to the modulation of junctional proteins of the cellular elements of the GFB and the tubular epithelial barrier.114,116,132 Rincon-Choles et al. 124 reported that exposure of glomerular cells to high glucose concentration results in decreased expression and serine/tyrosine phosphorylation of ZO-1 and its redistribution from the cell membrane to the cytoplasm. This translocation of ZO-1 is attenuated by an angiotensin-1 receptor blocker, indicating that this phenomenon is relevant to the pathogenesis of proteinuria in diabetic states. Xu et al. 133 demonstrated that high-glucose-treated podocytes in vitro reduced the P-cadherin mRNA levels and its protein content, with the involvement of protein kinase C, and the phenomenon is linked to the initial alterations of DN.
Recently, we showed that exposure of a renal tubular epithelial cell line, MDCK cells, to high glucose concentrations induced disruption of the paracellular barrier, as revealed by a decrease in transepithelial electrical resistance associated with an increased paracellular cation selectivity and permeability to molecules. 134 Immunocytochemical analyses showed a decreased junctional content of the barrier-forming claudins-1 and -3 as well as a significant increase in the junctional and cellular contents of pore-forming claudin-2, as well as time-dependent changes in the junctional content of occludin and ZO-1. These effects on the TJ-mediated epithelial barrier were independent of the osmotic changes caused by glucose, as indicated by the exposure of MDCK cells to mannitol, used at the same osmolality of the highest glucose concentration tested. 135 Indeed, the increase in osmolality had an opposite effect on the paracellular barrier to that observed for glucose. Thus, a significant increase in TEER (after 24 and 72 h) was observed after mannitol associated with a significant increase of claudin-1 junctional and cellular contents and a decrease in claudin-2 junctional and cellular contents. 135 Besides the effect of high glucose, exposition to high levels of FFAs (particularly the saturated palmitic acid) seems to have a deleterious effect on the TJ-mediated epithelial barrier in MDCK monolayers (unpublished data). Given the importance of the tubular epithelial barrier in renal transepithelial transport of ions and molecules, we suggest that its impairment caused by hyperglycemia and high FFAs levels would contribute to the renal dysfunction observed during DN.
Conclusions and perspectives
Taken all together, the IJs and their constitutive proteins seem to play an important role in the pathogenesis and complications of T2D, due to their involvement in both the compensatory hyperplasia process and in the secretory dysfunction of pancreatic beta cells, as well as in the impairment of the intestinal epithelial and glomerular/tubular barriers associated with diabetes mellitus (Figure 3). Also, one can foresee the participation of IJs in the T2D-associated impairment of the endothelial barrier, blood–retinal barrier, and blood–nerve barrier resulting in diabetic vascular disorder, retinopathy, and neuropathy, respectively.136–138 This knowledge opens the possibility that the junctional proteins can be used as target molecules in the gene, cell, and pharmacological therapy of T2D.
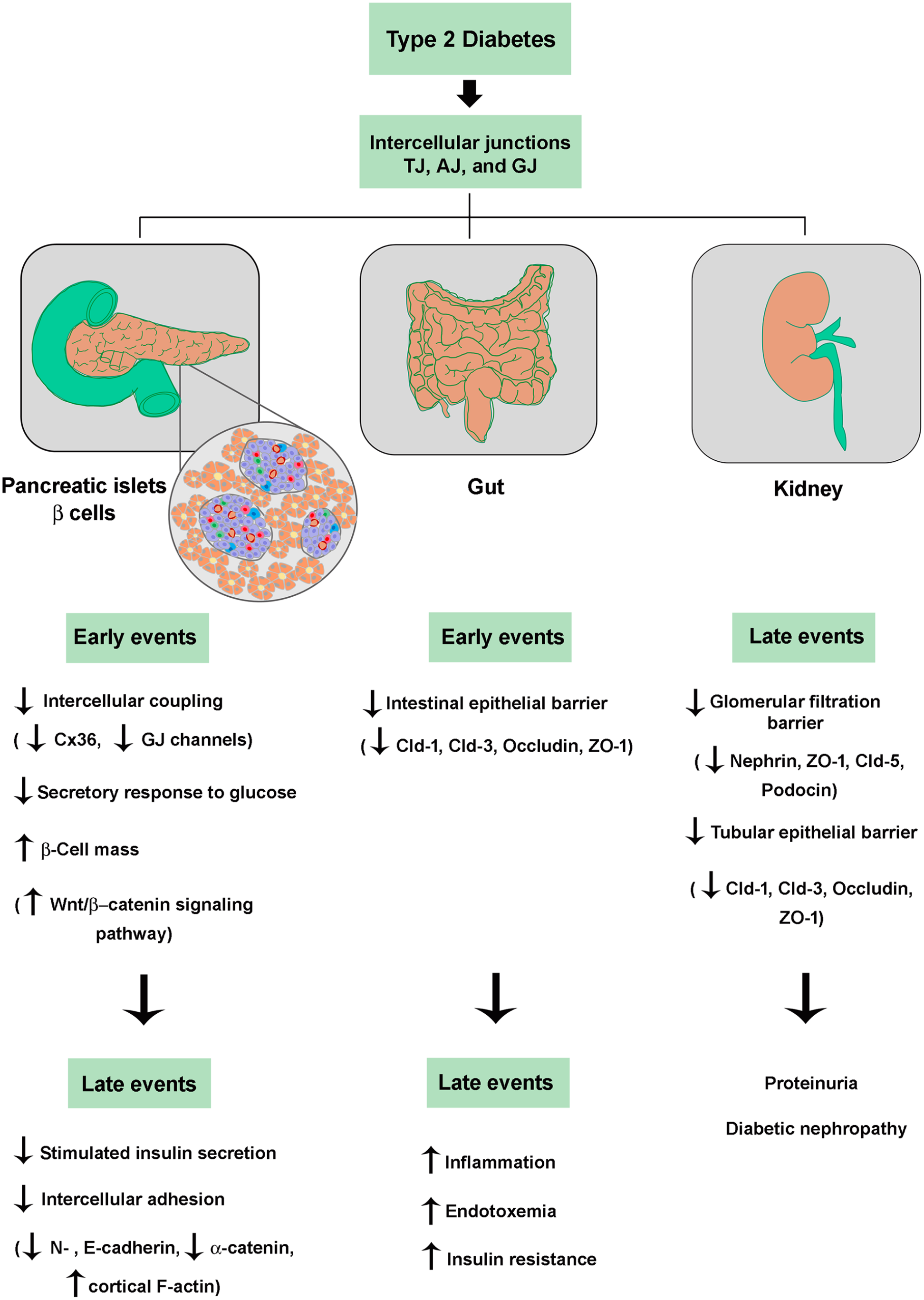
Role of intercellular junctions in dysfunctions of the endocrine pancreas (beta cells), gut, and kidney associated with pathogenesis and complications of T2D. See text for details. (A color version of this figure is available in the online journal.)
Footnotes
Acknowledgements
The authors thank Dr Marcos L Gazarini for the excellent help with the figure diagrams of this minireview and insights in the text.
Authors’ Contributions
Both the authors wrote and edited this manuscript and neither have any disclosures to report.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the São Paulo Research Foundation (FAPESP) (grant numbers 2010/50789-1, 2013/15676-0, 2014/22206-2, 2015/25442-1, and 2018/02118-2) and the Brazilian National Council for Scientific and Technological Development (CNPq) (grant numbers 201862/2007-7, 446957/2014-3, and 308546/2018-0).