
Abstract
Chronic kidney disease (CKD) is a high mortality disease and generally remains asymptomatic in the early stages. Long non-coding RNA (lncRNA) is defined as a non-protein-coding transcript more than 200 nucleotides which participate in numerous biological processes and have been identified as novel diagnostic markers for many diseases. Detection of circulating lncRNAs is a rapidly evolving, new area of molecular diagnosis. The purpose of our research was to identify circulating lncRNA expression profiles and possible molecular mechanisms involved in CKD. Blood samples were obtained from patients with CKD and healthy volunteers, and high-throughput sequencing was performed to identify differentially expressed (DE) lncRNAs and mRNAs. DE lncRNAs and mRNAs in peripheral blood mononuclear cells (PBMCs) were confirmed by quantitative reverse transcription polymerase chain reaction (qRT-PCR) to ensure the reliability and validity of RNA-seq data. Bioinformatics analysis was used to obtain biological functions and key pathways related to the pathogenesis of CKD. The interaction and co-expression functional networks for DE lncRNAs and mRNAs were also constructed. Our data showed that of the 425 DE lncRNAs detected, 196 lncRNAs were upregulated, while that of 229 lncRNAs were downregulated. A total of 433 DE mRNAs were identified in patients with CKD compared to healthy individuals. GO analysis revealed that DE lncRNAs were highly correlated with binding and pathway regulation. KEGG analysis suggested that DE lncRNAs were obviously enriched in regulatory pathways, such as antigen processing and presentation. We successfully constructed a potential DE lncRNA-mRNA co-expression network and analyzed the target genes of DE lncRNAs to predict cis- and trans-regulation in CKD. 100 lncRNAs that corresponded to 14 transcription factors (TFs) were identified in the TF-lncRNA binary network. Our findings on the lncRNA expression profiles and functional networks may help to interpret the possible molecular mechanisms implied in the pathogenesis of CKD; the results demonstrated that lncRNAs could potentially to be used as diagnostic biomarkers in CKD.
Keywords
Impact Statement
Chronic kidney disease (CKD) is recognized as a worldwide public health problem with a progressive increase in morbidity and mortality, and patients usually remain asymptomatic in the early stages. As an invasive method, renal biopsy is known as the gold standard for the diagnosis of CKD. Nowadays, noninvasive diagnostic methods for CKD may be of critical value. Circulating lncRNAs is a new area of molecular diagnosis for many diseases; however, none of the studies have reported the application of circulating lncRNAs for CKD diagnosis. The present study identified that 425 differentially expressed (DE) lncRNAs were associated with CKD, of these lncRNAs, for example, lnc-IFITM2-3 and lnc-FBXL5-7, may act as potential diagnostic biomarkers for CKD. Our findings also illustrated the expression profile and signature of circulating lncRNAs and their functional networks. The results could offer an improved understanding of lncRNAs involved in the pathogenesis of CKD, meanwhile, might serve as new noninvasive biomarkers.
Introduction
Chronic kidney disease (CKD) is characterized by progressive kidney damage or a decline in glomerular filtration rate (GFR), and associated with adverse outcomes, such as sarcopenia, anemia, and premature death. CKD is an escalating public health problem, with an estimated global prevalence of 8–16%,1–3 affecting 10.4% of men and 11.8% of women. 4 Approximately 1.2 million people died from CKD in 2016. 5 Patients usually remain asymptomatic in early stage, kidney biopsy is considered the gold standard in CKD diagnosis, but it is an invasive method. 6 Therefore, advanced noninvasive diagnostic methods for CKD are urgently needed to improve early diagnosis, guide medications, and evaluate disease progression.
Long non-coding RNA (lncRNA) is defined as a non-protein-coding transcript more than 200 nucleotides 7 that play a critical function in numerous biological processes, such as metabolism, cell cycle, and apoptosis. 8 Some studies have showed that lncRNAs with high sensitivity and specificity could be applied as diagnostic markers for various diseases. Li et al. 9 identified that lncRNA ENST00000444488.1 is a novel biomarker for the diagnosis of coronary artery disease. Cao et al. 10 demonstrated that lncRNA TUG1 might be a clinical diagnostic biomarker for systemic lupus erythematosus or lupus nephritis. Suwal et al. 11 reported that lncRNA NONRATT021972 is a potential diagnostic biomarker for diabetes-associated diseases. Moreover, researches have indicated that lncRNAs have a crucial function in modulating the occurrence, developmental process, and prognosis of renal fibrosis,12,13 implying that lncRNA may be an effective clinical biomarker for predicting CKD risk.
The secondary structures of lncRNAs are displayed highly stable, which makes quantitative analysis of body fluid samples feasible. Identification of circulating lncRNA in the peripheral blood is a rapidly evolving, new area of molecular diagnosis. However, to date, none of the studies have reported the application of circulating lncRNAs for CKD diagnosis. In the present study, we conducted high-throughput sequencing and bioinformatics analysis to identify the signature of circulating lncRNAs in blood of patients with CKD. The expression profiles and functional networks of these lncRNAs were examined to elucidate possible molecular mechanisms underlying CKD progression.
Materials and methods
Sample collection
Three patients with CKD and 3 healthy volunteers aged >18 years were recruited from Xinhua Hospital between October and November in 2019. All patients were diagnosed with non-dialysis CKD stage 5, and estimated GFR was calculated by simplified modification of diet in renal disease (MDRD) study equation. 14 Patients with diabetes mellitus, tumors, or undergoing immunosuppressant treatment were excluded; the background features of participants are presented with Supplemental Table S1. We drawn 8 ml fasting venous blood, centrifuged, isolated plasma, and stored at -80 °C. The ethics committee of our hospital approved this study.
RNA extraction and RNA library construction
MirVana miRNA isolation kit (Ambion, USA) was applied to isolate the total RNA. After assessing the integrity of RNA, the samples which RIN ⩾ 7 were selected to build RNA libraries. Based on these libraries, sequencing was performed on an Illumina sequencing platform (HiSeqTM 2500), generating 150/125 bp paired-end reads.
Analysis of DE lncRNAs and mRNAs
The DESeq software (R package) 15 was employed to normalize the lncRNA and mRNA counts for each sample, calculate the difference multiple, test the significance of the reads for negative binomial distribution, and find the expression differences between the lncRNAs and mRNAs.
PBMC preparation and qRT-PCR
Fasting blood samples (5 ml) from CKD patients and healthy participants were collected in ethylenediaminetetraacetic acid (EDTA) tubes. Ficoll–Paque density gradient centrifugation (Cytiva, Marlborough, American) was used to isolate peripheral blood mononuclear cells (PBMCs). Total RNA from PBMCs was extracted by TRIzol reagent (TianGen, Beijing, China). We conducted NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, USA) to measure the concentration of RNA. cDNA was synthesized using the RT reagent kit with gDNA Eraser (TaKaRa, Japan), and quantitative reverse transcription polymerase chain reaction (qRT-PCR) was used to confirm the reliability and validity of the RNA-seq data. The primer details are shown in Table 1.
Primer sequences for qRT-PCR.

Functional enrichment analysis
GO database was used to construct significant annotations of genes and gene products in this study. KEGG pathway analysis was performed for the DE lncRNAs in different biological pathways. Results with p value < 0.05 were considered statistically significant in functional enrichment analysis, and the top 20 of each analysis were extracted for visualization.
lncRNA-mRNA co-expression network
To identify the correlation between DE lncRNAs and mRNAs, a co-expression network of DE lncRNA-mRNA was built, and the expressed correlation was calculated with a Pearson correlation coefficient ⩾0.8 and a statistically significant p value < 0.05 to identify target genes.
LncRNA cis- and trans-regulation prediction analyses
As defined in previous researches, cis-regulators act on adjacent genes located on the same chromosome and lncRNAs was found to modulate gene expression in a cis-manner.16,17 The cis-regulatory regions were identified using a two-step approach: (1) The coding gene is located within the 100k that were upstream and downstream of a candidate lncRNA, and (2) the coding gene is significantly co-expressed with the candidate lncRNA (p value < 0.05). In terms of trans-regulation prediction, we used RIsearch2 to predict the binding site of a candidate lncRNA-gene; then, we concentrated on the lncRNAs that exert regulatory functions through transcription factors (TFs) and constructed TF-lncRNA- and TF-lncRNA-gene networks.
Statistical analysis
DESeq R package was performed to identify DE lncRNAs and mRNAs; GraphPad Prism 5.0 was used for statistical analyses; all results are expressed as mean ± standard error of the mean (SEM). Two-tail unpaired Student’s t-test was used to analyze statistical significance of randomly selected DE lncRNAs and mRNAs, while p value < 0.05 was considered statistically significant.
Results
Identification of DE lncRNAs and mRNAs by using RNA-Seq analysis
To identify whether circulating lncRNAs are differentially expressed in patients with CKD, we performed RNA-Seq analysis on blood samples obtained from patients with CKD and healthy volunteers. 24,577 lncRNAs were identified in the CKD group compared with the control group; we found 425 DE lncRNAs, 196 lncRNAs were upregulated whereas 229 lncRNAs were downregulated. The DE lncRNAs were depicted using a cluster heatmap (Figure 1(a)) and volcano plots (Figure 1(b)), respectively. We performed the lncRNA subgroup analysis to predict the biological function. As shown in Figure 2(a), these lncRNA transcripts are extensively located on the most chromosomes. DE lncRNAs were classified according to the level of direction, type, and location (Figure 2(b)). In addition, when the total 395,286 coding transcripts were examined, 433 mRNAs were found to exhibit significant differential expression profiles in patients with CKD compared to healthy volunteers; both cluster heatmap and volcano plots showed that 186 mRNAs were upregulated and 247 mRNAs were downregulated (Figure 1(c) and (d)).
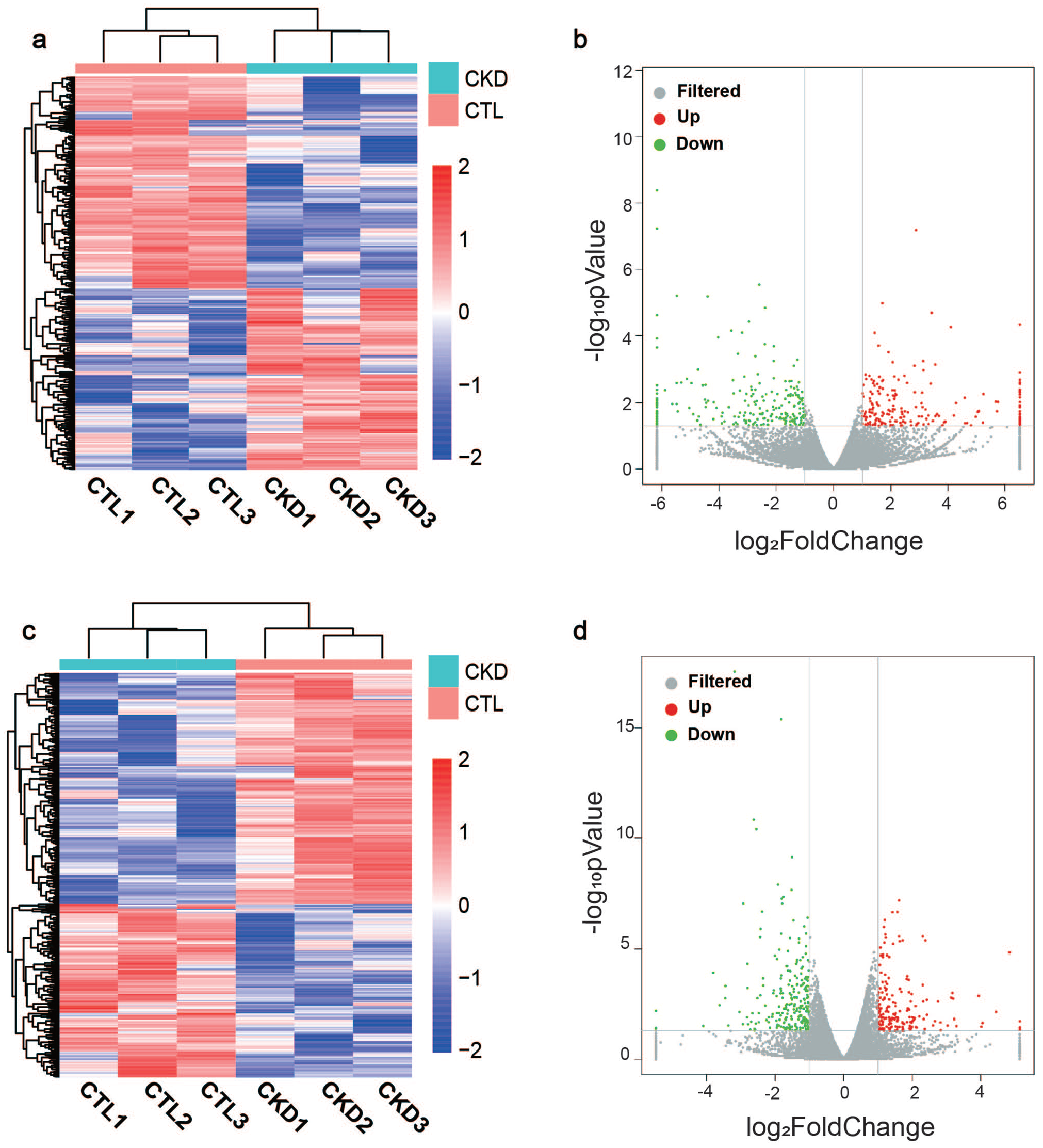
Differential expression of lncRNAs and mRNAs detected using RNA-Seq analysis: (a) The cluster heatmap showing DE lncRNAs in patients with CKD compared to healthy volunteers. (b) The volcano plots indicating all DE lncRNAs. Red and green represent upregulated and downregulated genes, respectively. (c) The cluster heatmap showing all DE mRNAs in patients with CKD compared to healthy volunteers. (d) The volcano plots indicating all DE mRNAs. (A color version of this figure is available in the online journal.)
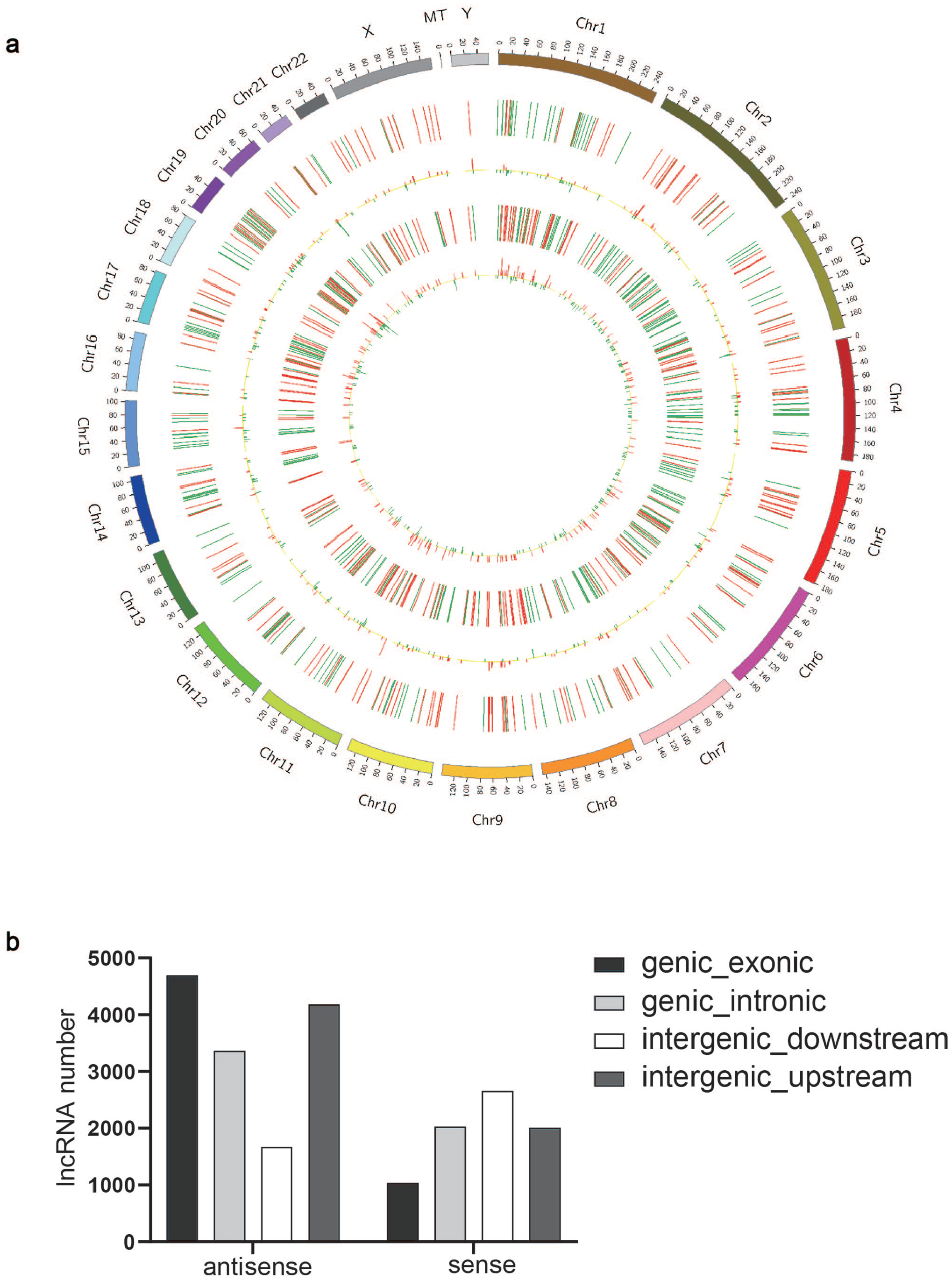
The distribution and classification of DE lncRNAs: (a) The distribution of DE lncRNAs was shown by the circos plot. (b) DE lncRNAs are classified according to the level of direction, type, and location. (A color version of this figure is available in the online journal.)
Verification of DE lncRNA and mRNA profiling by qRT-PCR
We randomly selected 5 DE lncRNAs to confirm the results of RNA-seq using qRT-PCR; our results showed that lnc-IFITM2-3, lnc-FBXL5-7, and lncRNA XR_930957.2 were upregulated in PBMCs of patients with CKD, whereas ENST00000358234 and ENST00000604749 were downregulated when compared to the control group (Figure 3(a)). We also randomly selected 5 common mRNA to evaluate gene expression and confirmed that CCR7, MMD, and MAP3K7CL were significantly downregulated, whereas ADGRE1 and ANXA1 were upregulated in PBMCs of patients with CKD (Figure 3(b)). We found the results of qRT-PCR analysis were highly consistent with the RNA sequencing, implying the reliability and validity of RNA-seq data in CKD.
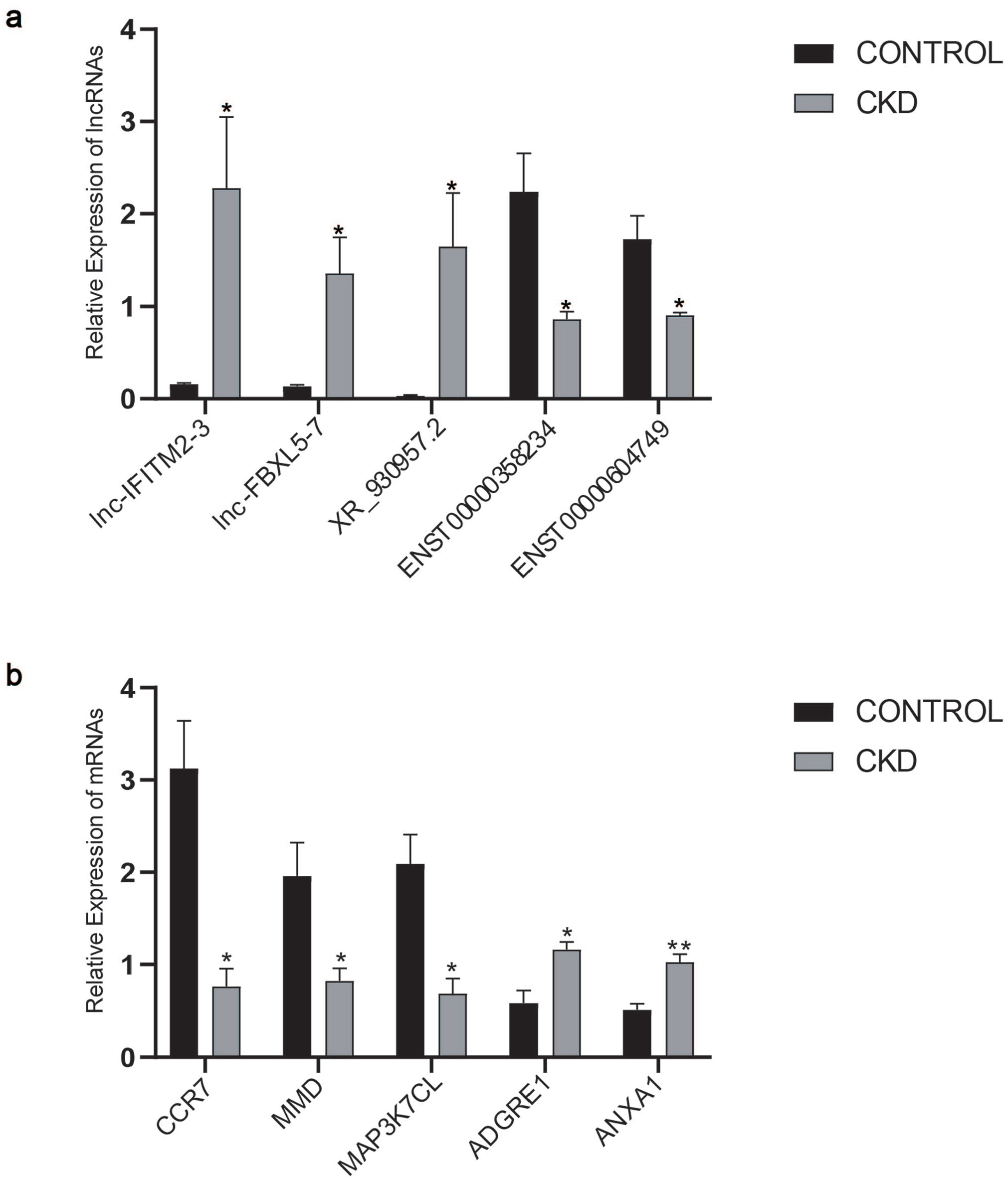
Verification of DE lncRNA and mRNA profiling by qRT-PCR: (a) The difference of DE lncRNAs between CKD and healthy volunteers (*p value < 0.05). (b) The difference of DE mRNAs between CKD and healthy volunteers (*p value < 0.05, ** p value < 0.01).
GO analysis and KEGG pathway analysis
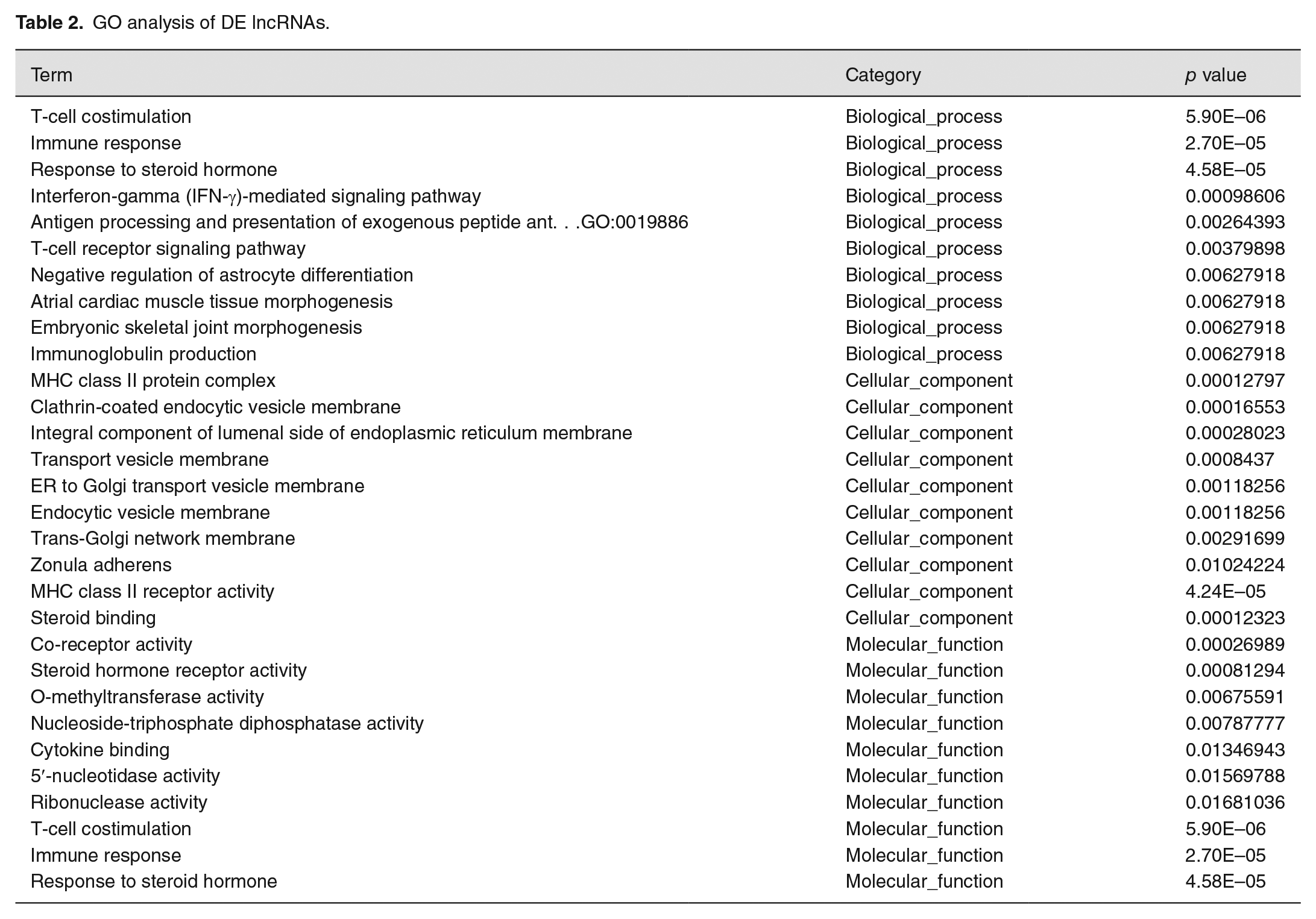
GO analysis on the DE lncRNAs revealed that the top 10 enriched GO terms were associated with biological processes, cellular components, and molecular function (Figure 4(a) and Table 2). In biological process domain, the top three enriched GO terms were T-cell co-stimulation, immune response, and interferon-gamma (IFN-γ)-mediated signaling pathway. The top three enriched GO terms in the cellular component domain were MHC class II protein complex, clathrin-coated endocytic vesicle membrane, and integral component of the lumenal side of the endoplasmic reticulum membrane. MHC class II receptor activity, O-methyltransferase activity, and nucleoside-triphosphate diphosphatase activity were the top three enriched GO terms in the molecular function domain.
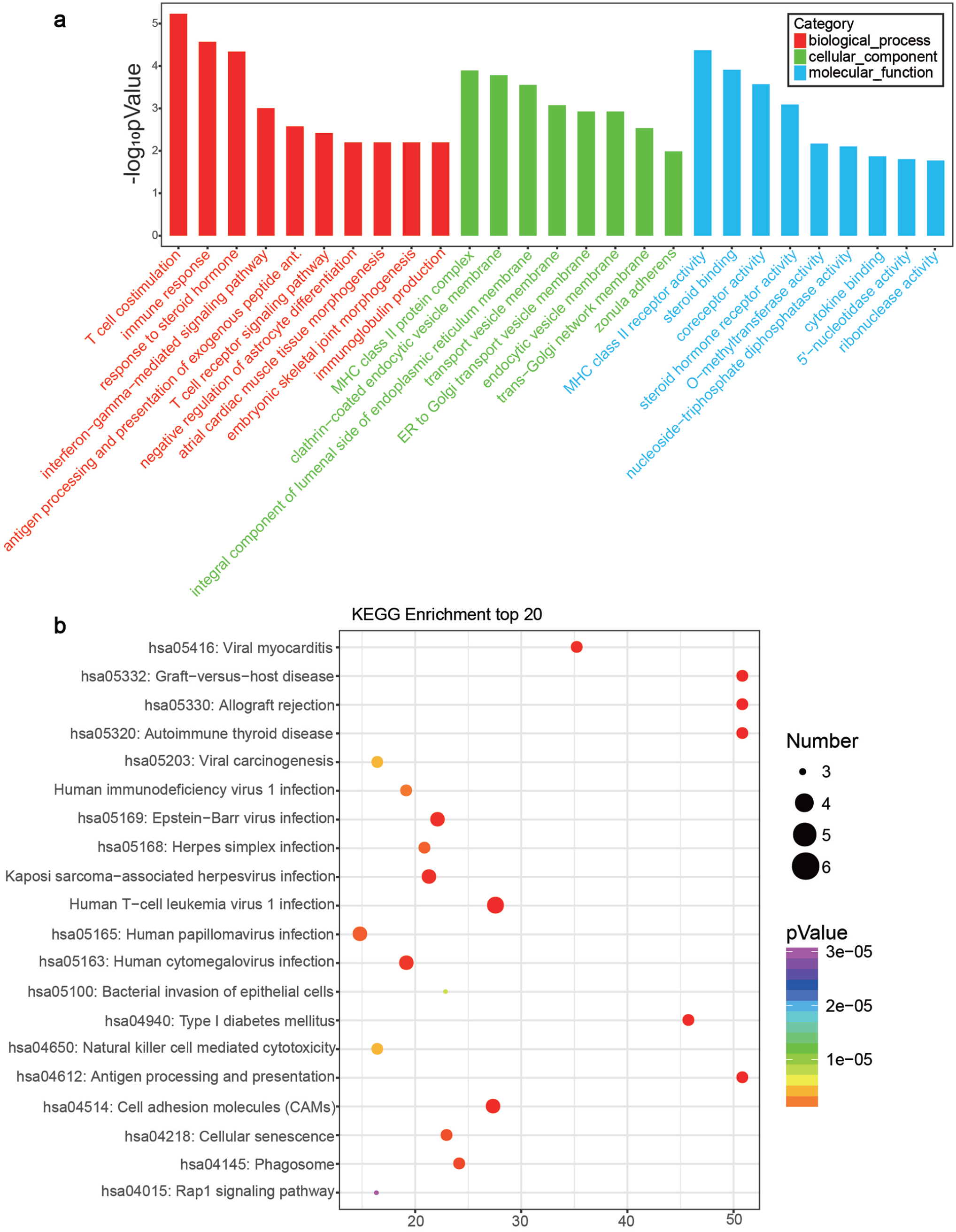
GO analysis and KEGG pathway analysis: (a) GO analysis showing the top 10 enriched terms. (b) KEGG pathway analysis displaying the top 20 enriched pathways. (A color version of this figure is available in the online journal.)
GO analysis of DE lncRNAs.
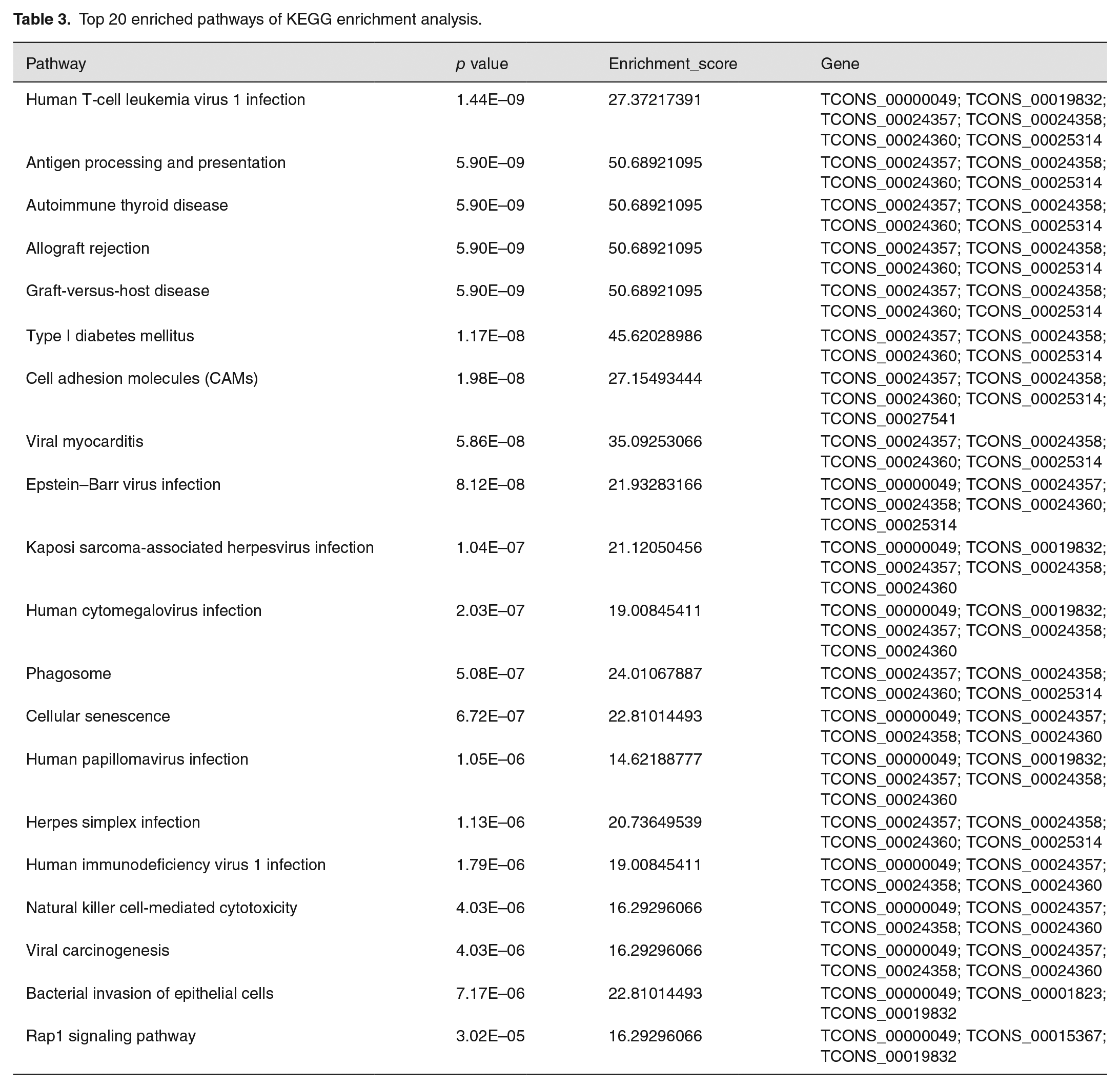
KEGG pathway analysis of DE lncRNAs showed that many pathways were significantly altered in patients with CKD. We found that the most affected pathways including cell adhesion molecules (CAMs), natural killer cell-mediated cytotoxicity, antigen processing and presentation, and Rap1 signaling pathway (Figure 4(b)). The corresponding p values and enrichment scores of the top 20 enriched pathways are shown in Table 3.
Top 20 enriched pathways of KEGG enrichment analysis.
Construction of mRNA-lncRNA co-expression network
To examine the complex relationship between DE lncRNAs and mRNAs, we constructed a co-expression network. Five upregulated and 5 downregulated lncRNAs associated with multiple biological processes were selected. As shown in Figure 5, upregulated lncRNA XR_ 001752103.1 regulates genes TEC and ORM1, whose mRNAs are implicated in autoimmune regulation. In addition, downregulated RGMA, which participated in the modulation of cytokine TGF and cellular metabolism, was found to interact with downregulated lncRNA XR_001753371.1 and upregulated lncRNA XR_001748284.1. The co-expression network implied a potential regulatory mechanism in the pathogenesis of CKD.
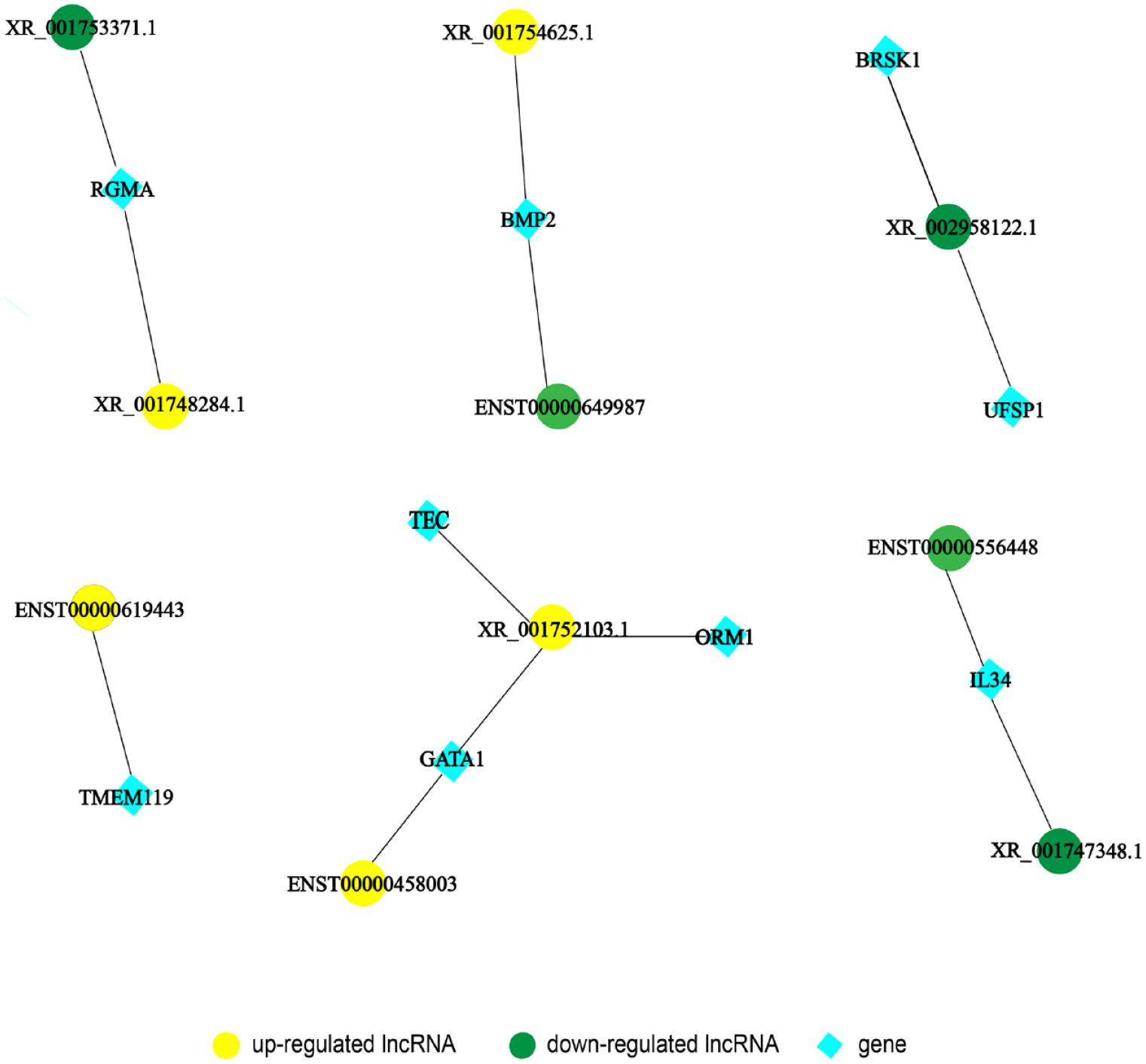
Construction of lncRNA-mRNA co-expression network. (A color version of this figure is available in the online journal.)
Cis- and trans-regulation prediction of lncRNAs
To understand the cis-regulatory function of lncRNAs, we predicted the relationship between DE lncRNAs and co-expressed adjacent coding genes. Since the cis of an lncRNA acts on neighboring target genes, we searched within the 100k that were upstream and downstream of the 425 DE lncRNAs to identify coding genes and analyzed their functions. Thirty-three lncRNAs were detected with adjacent protein-coding genes, with some having only a coding gene nearby, for instance, lncRNA XR_926755.2 and PNPLA1; lncRNA NR_038415.1 and RNF43; lncRNA ENST00000623759 and PIM3; lncRNA XR_936050.2 and ZNF610; and lncRNA XR_947722.2 and TSPAN2. Some lncRNAs were found to regulate two nearby coding genes, for example, lncRNA XR_001754625.1 regulates PI3 and SEMG1; lncRNA TCONS_00021252 regulates P2RY14 and P2RY12; lncRNA XR_938875.2 regulates PPBP and CXCL5; lncRNAs ENST00000524824, XR_930957.2, ENST00000531076, and XR_930956.1 simultaneously regulate IFITM2 and IFITM3, as indicated in the putative cis-regulatory network shown in Figure 6. Some genes are regulated by two lncRNAs simultaneously; for example, MUC20 is regulated by lncRNA ENST00000594101 and ENST00000414625; OXNAD1 is regulated by lncRNA ENST00000607464 and ENST00000606713, and CXCL5 is regulated by lncRNA XR_938875.2 and XR_938876.2. These networks may provide reference clues for the interaction between lncRNAs with nearby coding genes.
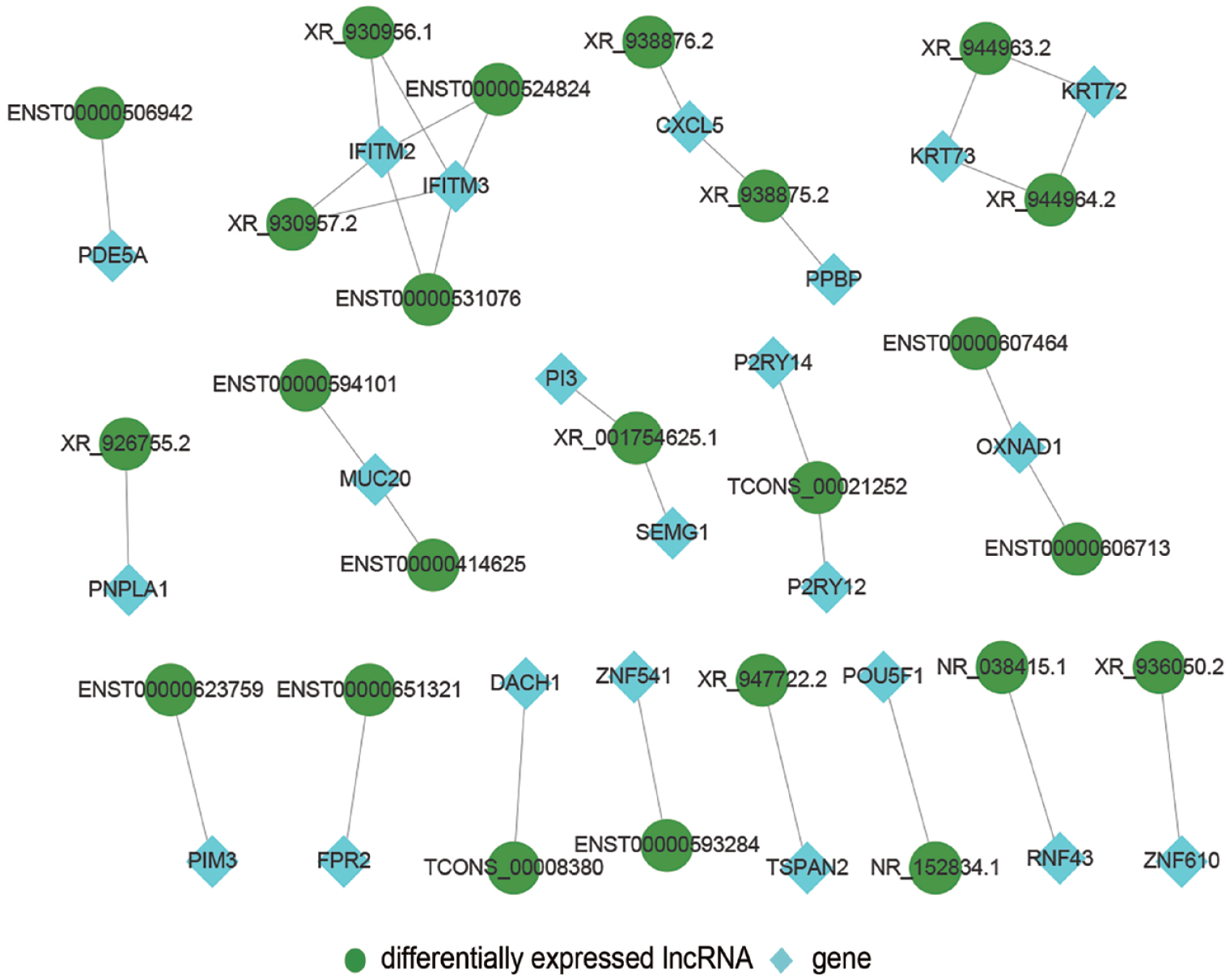
Predicted cis-regulation of lncRNAs: The network displays putative cis-regulation. (A color version of this figure is available in the online journal.)
To predict trans-regulation, a co-expression network combining these DE lncRNAs and TFs was constructed. Under the thresholds of p value < 0.01, we selected the top 200 closest TFs, and the potential trans-regulatory network is shown in Figure 7. We also selected 100 lncRNAs, which were related to 14 TFs to build a TF-lncRNA binary network (Figure 8). In addition, we introduced target genes to construct a TF-lncRNA-gene ternary network, as shown in Figure 9, with 10 lncRNAs corresponding to 1,096 genes through five TFs, including LEF1, MIER2, FOXH1, ZNF586, and FDX1. Our findings suggested that these TFs might participate in the pathogenesis of CKD.

Predicted trans-regulation of lncRNAs: The network displays putative trans-regulation. (A color version of this figure is available in the online journal.)
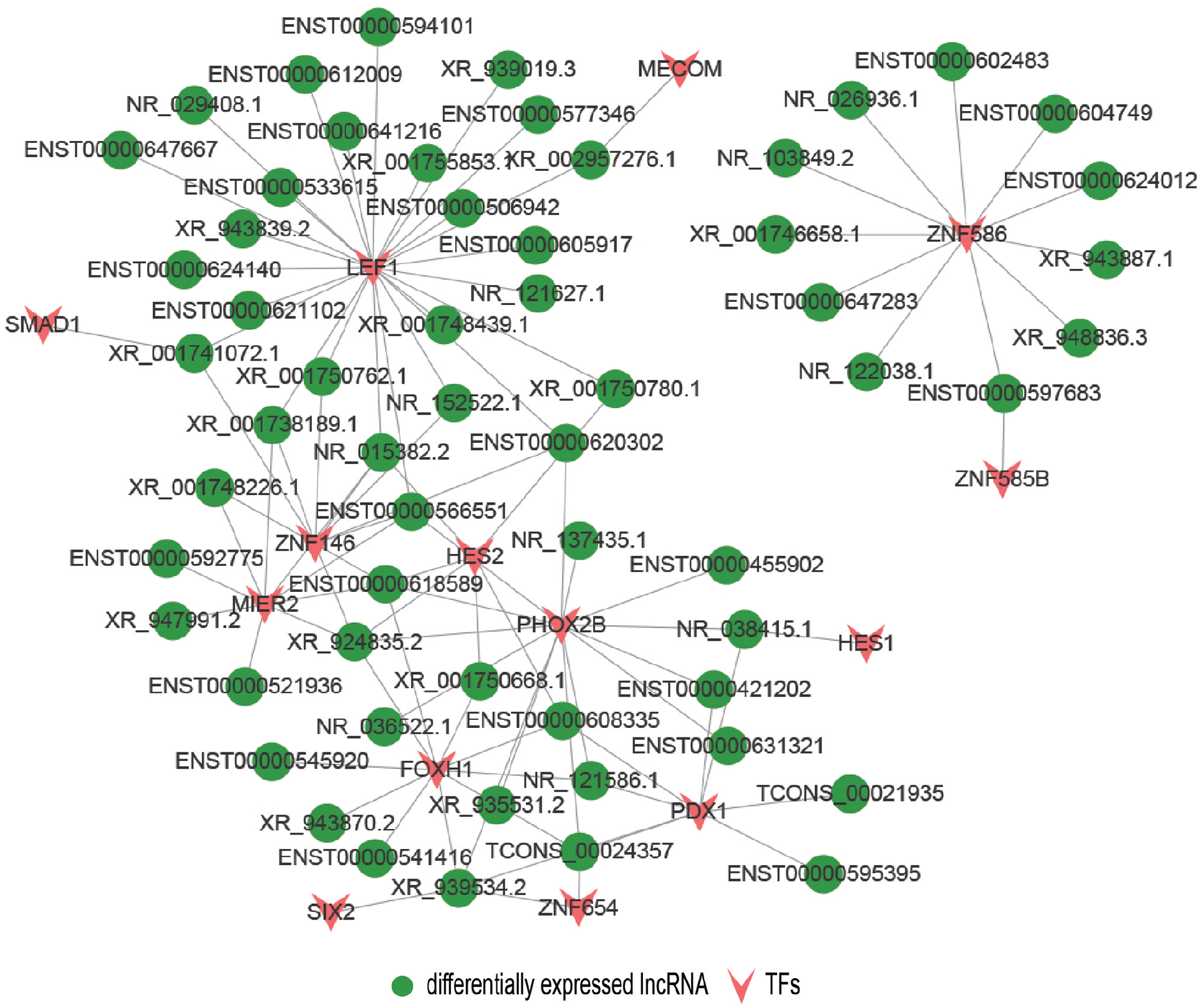
TF-lncRNA binary network showing 100 DE lncRNAs corresponded to 14 TFs. (A color version of this figure is available in the online journal.)
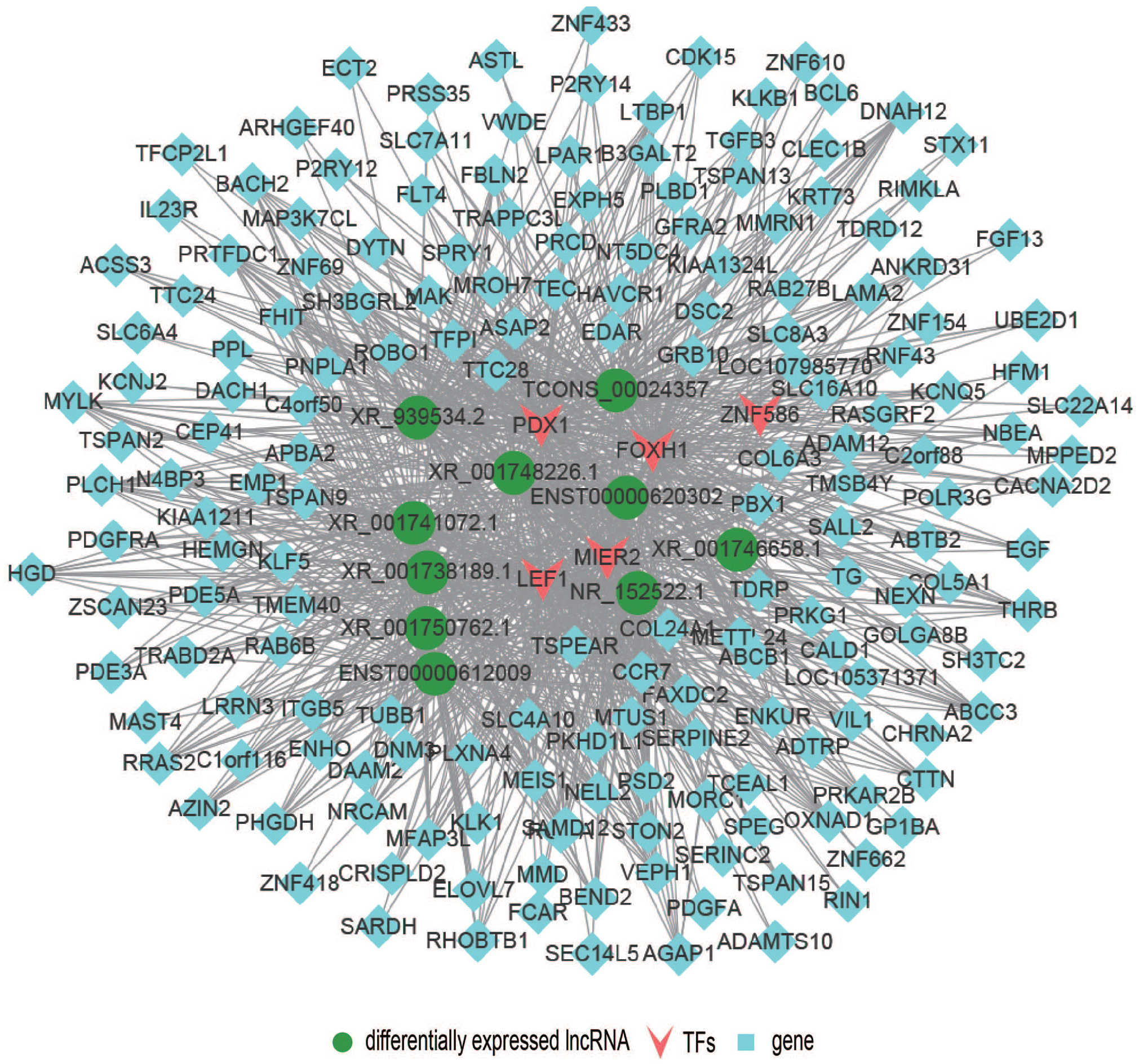
TF-lncRNA-gene ternary network showing 10 lncRNAs corresponded to 1,096 target genes via 5 transcription factors. (A color version of this figure is available in the online journal.)
Discussion
CKD is asymptomatic in the early stages, and less than 5% patients with CKD are diagnosed.18,19 Although an invasive method, renal biopsy remains the gold standard for the CKD diagnosis. However, with the rapid development of diagnostic technology and precision medicine, studies on devising noninvasive bioinformatics methods are urgently needed. Circulating lncRNAs in the peripheral blood, which have a highly stable secondary structure, is potential novel molecular diagnostic markers for many diseases. In 2019, Zhao et al. 20 reported that the circulating lncRNA BACE1-AS may be a potential marker for Alzheimer’s disease. Similarly, Yuan et al. 21 found that circulating lncRNA LINC00152 could be used to diagnosis hepatocellular carcinoma.
Circulating lncRNAs is a new area of molecular diagnosis for many diseases; however, none of the studies have reported the application of circulating lncRNAs for CKD diagnosis. In our current study, high-throughput RNA-seq on the plasma samples was performed, and we identified many circulating lncRNAs including 425 DE lncRNAs and 433 DE mRNAs that were strongly associated with CKD progression, the reliability and validity of the RNA-seq data were also verified by qRT-PCR result. To elucidate the possible molecular mechanisms, GO analysis and KEGG pathway analysis were performed. Our results showed that the top 10 enriched GO biological processes associated with DE lncRNAs included T-cell costimulation, immune response, and IFN-γ-mediated signaling pathway. Some of the most enrichment KEGG signaling pathways, including antigen processing and presentation, CAMs, natural killer cell-mediated cytotoxicity, and Rap1, are shown to be associated with the progression of CKD. Poosti et al. 22 reported that IFN-γ could inhibit fibroblast activation and proliferation, and targeted delivery of IFN-γ might reduce the expression of nephrocolloid I, interstitial T-cell immersion, and lymphatic tube production, thereby attenuating fibrosis in unilateral ureteral obstruction (UUO) mice. Studies have also confirmed that the IFN-γ-mediated signaling pathway acts a crucial role in renal fibrosis.23–25 Zuk and Bonventre 26 reported that the development of kidney disease is driven by chronic inflammatory process; changes in protein folding and mitochondrial function can trigger various disorders such as impaired immune response and upregulated inflammation, thereby destroying the tissue structure and functions of the kidney. Our results of GO/KEGG pathway analysis revealed that many DE lncRNAs and enriched pathways may be related to the progression of CKD, which is consistent with previous findings.
To further elucidate the molecular mechanisms of CKD, we successfully constructed a potential DE lncRNA-mRNA co-expression network. Such networks are huge and complex, and each component in the network can be remarkably related. Although this co-expression network implies a potential lncRNA-mRNA regulatory mechanism in the pathogenesis of CKD, research data on this relationship remain limited.
LncRNAs regulate co-expression of nearby coding genes via cis-regulation. 16 In this study, we searched for all coding genes within the 100k that were upstream and downstream of the DE lncRNAs and constructed a cis-regulated network. We also confirmed 33 lncRNAs have adjacent protein-coding genes; some lncRNAs only have one nearby coding gene, such as lncRNA NR_038415.1 and RNF43. Chan et al. 27 found that RNF43 regulates the expression of hepatocyte nuclear factor-1β that regulates renal tubular function. In addition, some lncRNAs can regulate two nearby coding genes, for example, lncRNA XR_ 001754625.1 that regulates PI3 and SEMG1. Zhang et al. 28 reported that PI3 regulated kidney function and pathological changes in diabetic kidney disease through the INS/IRS-1/PI3K/Akt signaling pathway. Our results are consistent with the previous reports, revealing the functional cis-regulatory role of DE lncRNAs in CKD.
Although cis-regulating lncRNAs are more prevalent than trans-acting lncRNAs, 29 studies have shown that lncRNAs can bind to specific sites or sequences of TFs to exert their trans-regulatory functions. Using gene expression data, we constructed a co-expression network and identified 100 lncRNAs that corresponded to 14 TFs. We further constructed a TF-lncRNA binary network by introducing target genes and identified a total of 10 lncRNAs that corresponded to 1,096 genes via five TFs (FDX1, LEF1, MIER2, FOXH1, and ZNF586). Shang et al. 30 found that LEF1 can enhance the proliferation of renal cell carcinoma and may be a possible therapeutic target. Previous study has demonstrated that TGF-β can act as an important factor in the progression of renal fibrosis. 31 Liu et al. 32 further identified FOXH1 as a transcription regulator for Smads, which are the core element of the intracellular signaling pathway of TGF-β ligands, indicating that FOXH1 might have a key role in the development of renal fibrosis. Thus, cis- and trans-regulation predictions of our research may imply novel perspectives into the molecular mechanisms of CKD.
Our study had several limitations. The background characteristics of the CKD patients in our study are similar; for example, they all Chinese race, but CKD is a hugely heterogenous disease with contributions from genomic, racial, and environmental factors; thus, our outcomes may not be generalizable to other populations and races. In addition, our sample size is small which may probably affect the accuracy of the results, as we know a limited sample size may be insufficient to demonstrate significant differences. So, we plan to design a clinical trial with larger sample sizes to confirm these novel biomarkers.
In conclusion, our research was the first report to illustrate the expression profile and signature of circulating lncRNAs and their functional networks in patients with CKD. Our findings further elucidate the potential diagnostic value of lncRNA as a biomarker and a putative factor associated with the pathogenesis of CKD; these results suggest that circulating lncRNA may be used for rapid and noninvasive diagnosis of CKD in the future.
Supplemental Material
sj-docx-1-ebm-10.1177_15353702221104035 – Supplemental material for Identification of circulating lncRNA in chronic kidney disease based on bioinformatics analysis
Supplemental material, sj-docx-1-ebm-10.1177_15353702221104035 for Identification of circulating lncRNA in chronic kidney disease based on bioinformatics analysis by Rong Huang and Li-Jing Sun in Experimental Biology and Medicine
Footnotes
Authors’ Contributions
SLJ was responsible for the whole study and final version submission. HR contributed to analyze data, explain results, and write article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
The ethics committee of Xinhua Hospital Affiliated to Shanghai Jiaotong University, School of Medicine has approved this study.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The National Science Foundation of China (grant no. 81603558) and the Three-Year Plan for the Development of Chinese Medicine of the Shanghai Municipal Health Commission (grant no. ZWB-1001-CPJS40) have endorsed this study.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.