
Abstract
Chronic inflammation is a major contributor to the development of obesity-induced insulin resistance, which then can lead to the development of type 2 diabetes (T2D). Skeletal muscle plays a pivotal role in insulin-stimulated whole-body glucose disposal. Therefore, dysregulation of glucose metabolism by inflammation in skeletal muscle can adversely affect skeletal muscle insulin sensitivity and contribute to the pathogenesis of T2D. The mechanism underlying insulin resistance is not well known; however, macrophages are important initiators in the development of the chronic inflammatory state leading to insulin resistance. Skeletal muscle consists of resident macrophages which can be activated by lipopolysaccharide (LPS). These activated macrophages affect myocytes via a paracrine action of pro-inflammatory mediators resulting in secretion of myokines that contribute to inflammation and ultimately skeletal muscle insulin resistance. Therefore, knowing that synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acids (CDDOs) can attenuate macrophage pro-inflammatory responses in chronic disorders, such as cancer and obesity, and that macrophage pro-inflammatory responses can modulate skeletal muscle inflammation, we first examined whether CDDO-ethyl amide (CDDO-EA) inhibited chemokine and cytokine production in macrophages since this had not been reported for CDDO-EA. CDDO-EA blocked LPS-induced tumor necrosis factor-alpha (TNF-α), monocyte chemotactic protein-1 (MCP-1), interleukine-1beta (IL-1β), and interleukine-6 (IL-6) production in RAW 264.7 mouse and THP-1 human macrophages. Although many studies show that CDDOs have anti-inflammatory properties in several tissues and cells, little is known about the anti-inflammatory effects of CDDOs on skeletal muscle. We hypothesized that CDDO-EA protects skeletal muscle from LPS-induced inflammation by blocking nuclear factor kappa B (NF-κB) signaling. Our studies demonstrate that CDDO-EA prevented LPS-induced TNF-α and MCP-1 gene expression by inhibiting the NF-κB signaling pathway in L6-GLUT4myc rat myotubes. Our findings suggest that CDDO-EA suppresses LPS-induced inflammation in macrophages and myocytes and that CDDO-EA is a promising compound as a therapeutic agent for protecting skeletal muscle from inflammation.
Keywords
Impact statement
The significance of our findings is that it defines factors affected by 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-oic acids (CDDOs) to inhibit inflammation with the potential to protect from obesity-induced insulin resistance. We believe that our research will significantly advance the field of study in understanding the molecular mechanisms affected by CDDOs in skeletal muscle cells. The mechanistic effects of any CDDO in mitigating skeletal muscle inflammation related to insulin resistance have not been fully investigated. Our findings show that CDDO-EA protects skeletal muscle cells from activation of the pro-inflammatory NF-κB signaling pathway. Given the pivotal role of skeletal muscle in whole-body glucose homeostasis and its involvement in the pathogenesis of T2D, our investigation of how CDDO-EA alters inflammation in skeletal muscle could result in advancing the use of CDDOs as cost-effective therapeutic agents to treat and prevent obesity, insulin resistance, and T2D.
Introduction
Skeletal muscle is the major site of insulin-induced glucose uptake. 1 Insulin binds to its receptor on the plasma membrane, initiating a signaling cascade resulting in the translocation of GLUT4 to the plasma membrane. Insulin resistance in skeletal muscle is mainly due to impaired insulin signaling and glucose transport and is a hallmark of obesity and T2D. 1 The exact mechanism underlying insulin resistance in the skeletal muscle is not understood, but evidence indicates that inflammation occurs in skeletal muscle, similarly to adipose tissue.
In addition, it has been theorized that chronic inflammation may be a result of high circulating levels of lipopolysaccharide (LPS) that can result from a change in gut microflora associated with obesity. 2 Due to loss of integrity and increased permeability of the gut associated with obesity, LPS can more freely translocate from the intestinal lumen to the bloodstream resulting in chronically elevated plasma levels of LPS. 2 Elevated LPS plasma levels are found in insulin-resistant obese and type 2 diabetic individuals, and this is negatively correlated with skeletal muscle insulin sensitivity. 3 LPS is a ligand for Toll like receptor 4 (TLR4) and induces activation of the pro-inflammatory NF-κB signaling cascade through TLR4. 4 Our published studies demonstrated that insulin-resistant individuals have increased TLR4 content in the skeletal muscle. 5 In addition, our work showed that obese and T2DM individuals have increased NF-κB activation and interleukin (IL)-6 gene expression in their skeletal muscle. 5 Thus, a rise in LPS associated with obesity could activate NF-κB through TLR4 signaling in skeletal muscle that would result in a release of chemokines/cytokines from resident macrophages, initiating inflammation-induced insulin resistance.
Animal and human studies indicate that immune cells infiltrate skeletal muscle during obesity. 6 LPS may activate skeletal muscle–resident macrophages and induce a switch to the pro-inflammatory M1 macrophage phenotype. Activated M1 macrophages could then affect myocytes via paracrine action of pro-inflammatory mediators. In turn, myocytes might then secrete mediators that also contribute to inflammation. 7 Immune cells are recruited to skeletal muscle in response to MCP-1 produced by the skeletal muscle. Our published work demonstrated that MCP-1 production by muscle can exacerbate disease progression due to increased trafficking and activation of leukocytes in muscle. 7 After 1 week of a high-fat diet (HFD), recruitment of monocytes, macrophages, and neutrophils occurs in quadriceps of wild-type (WT) mice, while quadriceps of MCP-1 knockout mice on HFD had less macrophage recruitment and higher insulin sensitivity (increased Akt phosphorylation). 8 As a result, knowing that activation of immune cells, specifically macrophages, could adversely affect skeletal muscle inflammation resulting in the development of skeletal muscle insulin resistance, we wanted to examine whether CDDO-EA inhibited production of pro-inflammatory immune mediators in macrophages.
CDDOs are synthetic triterpenoid with modifications at the C17 position, such as methyl ester (Me), ethyl amide (EA), and imidazole (Im) groups. These modifications on C17 generate analogs of CDDO with varying pharmacological efficacies on biological effects and signaling pathways depending on the target cell type. 9 For example, although CDDO-Me, with methyl ester at C17, delayed the formation of estrogen receptor-negative mammary tumors in a mouse model of breast cancer, 10 CDDO-EA, with ethyl amide at C17, did not inhibit tumor formation in this same mouse model. Studies have also used various CDDOs in animal models of diabetes.11,12 Nonetheless, there has been little investigation of the effect of CDDOs on skeletal muscle. Given the pivotal role of skeletal muscle in whole-body glucose homeostasis and its involvement in the pathogenesis of T2D, an investigation of how CDDOs alter inflammation in skeletal muscle is warranted. One study demonstrated that CDDO-Me prevented inflammation in livers of mice fed a HFD as determined by macrophage infiltration and tumor necrosis factor (TNF)-α and IL-6 mRNA expression. However, skeletal muscle was not examined in that study. 12 Another study did investigate the effects of CDDO-EA and CDDO-trifluoroethyl amide (TFEA) on skeletal muscle. This study demonstrated that CDDO-EA and CDDO-TFEA reduced oxidative stress in skeletal muscle, improved motor impairment, and increased longevity in a mouse model of Huntington’s disease fed chow incorporated with either CDDO-EA or CDDO-TFEA. 13 In addition, CDDO-EA and CDDO-TFEA highly accumulated in the skeletal muscle of these mice who were fed chow incorporated with either CDDO-EA or CDDO-TFEA. Therefore, together these findings led us to further investigate whether CDDO-EA could block inflammation in skeletal muscle. In this investigation, we demonstrate that CDDO-EA inhibits LPS-induced inflammation in macrophages and myocytes and that the mechanism of inhibition is through NF-κB in muscle cells. Also, we show that CDDO-EA induces GLUT4 translocation and p38 phosphorylation in muscle cells. Our findings suggest that CDDO-EA may be suitable as a therapeutic agent for protecting skeletal muscle from inflammation.
Materials and methods
Reagents
CDDO-EA was a kind gift from Dr. Thomas J. Slaga, PhD, Department of Pharmacology, The University of Texas Health Science Center at San Antonio. Dr. Slaga received CDDO-EA from Dr. Michael M. Sporn from Dartmouth Medical School laboratory where CDDO-EA’s purification was verified. CDDO-EA was dissolved in DMSO (Sigma Aldrich) and stored in 40 mM stock concentrations at −80°C until used for experiments. Vehicle controls contained concentration of 0.125% DMSO.
Cell culture
The L6-GLUT4myc rat myoblast cell line was kindly provided by Dr. Amira Klip, Hospital for Sick Children, Toronto, Ontario, Canada. L6-GLUT4myc myoblasts were maintained and differentiated to myotubes as previously described. 14 L6-GLUT4myc rat myoblast cells were not used beyond passage 11.
Mouse RAW 264.7 macrophage cells were cultured as described previously. The cells were then sub-cultured using 5 mL of ice cold 5 mM EDTA in PBS in 4°C for 20 min, tapping the flask every 5 min to lift the cell from the flask. The cell pellet was collected by centrifugation at 800 r/min for 5 min at room temperature.
THP-1 human monocytes were maintained in culture as previously described. 15 THP-1 monocytes were sub-cultured when the cell concentration reached 8 × 105 cells/mL. To differentiate THP-1 monocytes to macrophages, THP-1 monocytes were seeded at 1 × 106 cells/mL in 6-well Corning® cell culture treated dishes in growth medium + 100 ng/mL PMA (phorbol 12-myristate 13-acetate) and cultured at 37°C with 5% CO2 incubator for 48 h to achieve differentiation to macrophages. 15
Western blotting and subcellular fractionation
L6-GLUT4myc myoblasts (4 × 104 cells/mL, 2 mL/well) were seeded onto 6-well dishes and differentiated with differentiation medium for 7 days. Myotubes were treated with LPS 100 ng/mL 16 (Escherichia coli O111: B4, Sigma Aldrich) in the absence or presence of CDDO-EA 500 nM, as indicated. After treatment, myotubes were lysed using lysis buffer (containing 1% Triton-X) with 50 mM Hepes pH 7.6 (Sigma Aldrich), 50 mM NaCl (Sigma Aldrich), 20 mM sodium pyrophosphate (Thermo Fisher Scientific), 20 mM β-glycerophosphate (Sigma Aldrich), 10 mM NaF (Thermo Fisher Scientific), 1 mM Na3VO4 (Sigma Aldrich), 1 mM PMSF (Sigma Aldrich,), 1 mM aprotinin (Sigma Aldrich) and 1 mM leupeptin (Sigma Aldrich). Protein concentrations were determined using the Pierce BCA Protein Assay (Thermo Fisher Scientific), and equal amounts of protein were separated by electrophoresis and subsequently transferred to nitrocellulose membranes. Membranes were analyzed using the following antibodies from Cell Signaling Technology: phosphorylated NF-κB (Ser536) (93 H1, cat# 3033,1:1000), phosphorylated IκB-α (Ser32) (14D4, cat# 2859,1:1000), IκB-α (44D4, cat# 4812, 1:1000), phosphorylated p38 (Thr180/Tyr182) (cat # 9211, 1:1000), p38 (cat # 9212, 1:1000). Membranes were also analyzed using NF-κB p65 (C-20, cat# sc-372, 1:1000) and Lamin A/C (H-110, cat# sc-20681, 1:1000) from Santa Cruz Biotechnology and actin antibody (cat# A1978, 1:5000) from Sigma. Primary antibodies were detected using donkey anti-rabbit (cat# sc-2077, 1:5000) or donkey anti-mouse (cat# sc-2314, 1:5000) secondary antibodies conjugated to horseradish peroxidase from Santa Cruz Biotechnology. Bands were quantitated using ImageQuant (GE Healthcare). Phosphorylated protein was normalized to total protein.
For subcellular fractionation, differentiated L6-GLUT4myc myotubes were pre-treated with CDDO-EA 500 nM for 1 h follow by LPS 100 ng/mL 16 (E. coli O111: B4, Sigma Aldrich) treatment for 1 h, and the nuclear and cytosolic fractions were isolated using the Nuclear Extraction Kit (Panomics) according to the manufacturer’s instructions. The extracted fractions were then analyzed by Western blotting as described. 15
Immunofluorescence microscopy
L6-GLUT4myc myoblasts (4 × 104 cells/mL, 0.2 mL/well) were plated onto Nunc Lab-TekII 8-chamber slides (Thermo Fisher Scientific) and differentiated for 7 days. Myotubes were pre-treated with CDDO-EA 500 nM for 1 h followed by LPS 100 ng/mL 16 (E. coli O111: B4, Sigma Aldrich) treatment for 1 h. After treatment, myotubes were fixed, permeabilized, and stained with anti-NF-κB p65 primary antibody (cat# sc-372, 1:100, Santa Cruz Biotechnology) and a goat anti-rabbit secondary antibody conjugated with Alexa Fluor® 488 (cat# A32731, 1:500, Invitrogen) as previously described. 15 For GLUT4myc cell membrane staining, myotubes were pre-treated with CDDO-EA 500 nM for 1 h followed by LPS 100 ng/mL 16 (E. coli O111: B4, Sigma Aldrich) treatment for 1 h. Insulin 100 nM (cat # I9278, Sigma Aldrich) treatment was for 15 min. After CDDO-EA, LPS, and insulin treatment, myotubes were fixed without permeabilization and stained with anti-c-myc primary antibody (cat# C3956, 1:100, Sigma Aldrich) and a goat anti-rabbit secondary antibody conjugated with Alexa Fluor 488 (cat# A32731, 1:500, Invitrogen) as previously described. 15
Luciferase assay
L6-GLUT4myc myoblasts were transfected with 2 μg of phosphorylated NF-κB-MetLuc2-Reporter plasmid as described previously. After transfection, cells were plated onto a 24-well dish (1.5 × 105 cells/well) and pre-treated with CDDO-EA for 1 h. Myotubes were then stimulated with LPS 100 ng/mL 17 (E. coli O111: B4, Sigma Aldrich) for 6 h, and luciferase activity was measured using the Ready-To-GlowTM NF-κB Secreted Luciferase Reporter System as described. 15
Quantitative RT-PCR
L6-GLUT4myc myoblasts (4 × 104 cell/mL, 2 mL/well) were seeded on a 6-well plate and differentiated into myotubes with differentiation medium for 7 days. Myotubes were pre-treated with CDDO-EA 500 nM for 1 h followed by LPS 100 ng/mL 16 (E. coli O111: B4, Sigma Aldrich) treatment for 1 h. RNA was isolated from treated myotubes using TRIzol reagent (Ambion) according to the manufacturer’s instructions. Validated RT-PCR primers specific for rat TNF-α (Assay ID: Rn99999017_m1) and rat MCP-1 (Assay ID: Rn00580555_m1), or rat GAPDH (Assay ID: Rn99999916_s1) were used to quantify mRNA levels using TaqMan RNA-to-CT 1-Step Kit (Thermo Fisher Scientific), Real-time PCR was carried out using an AB7900HT Fast Real-Time PCR System (Applied Biosystems), and MCP-1 and TNF-α levels were calculated using the ∆∆CT method according to the manufacturer’s instructions.
Cytokine detection
RAW 264.7 cells were grown on 24-well dishes at 1 × 106 cells/mL, 0.5 mL/well. Cells were pre-treated with CDDO-EA 500nM for 1 h. After 1 h, cells were treated with 100 ng/mL of LPS16,18 (E. coli O111: B4, Sigma Aldrich) for 6 h, and supernatants were collected and stored at −80°C until analysis. TNF-α from the culture media of cells was measured and quantified using a Mouse TNF (Mono/Mono) ELISA Set (BD Bioscience) according to the manufacturer’s instructions.
THP-1 monocytes were plated in 24-well plate at 15 × 104 cells/0.5 mL/well in growth medium plus 100 ng/mL PMA into each well and cultured at 37°C with 5% CO2 incubator for 48 h for differentiation to macrophages. After 48 h, THP-1 macrophages were pre-treated with CDDO-EA 500 nM for 1 h followed by LPS 100 ng/mL15,17 (E. coli O111: B4, Sigma Aldrich) exposure for 6 h. Supernatants were collected and stored at −80°C until analysis. Cytokines and chemokines were measured and quantified using a Bio-Plex Pro human cytokines group I, 17-plex ELISA Kit (Bio-Rad) according to the manufacturer’s instructions.
Sulforhodamine B (SRB) cell viability assay
L6-GLUT4myc myoblasts (4000 cells/well) were seeded on a 96-well plate and differentiated into myotubes with differentiation medium for 7 days. Myotubes were then treated with doubling concentrations of CDDO-EA for 6 or 24 h. After treatment, myotubes were fixed in 10% cold trichloroacetic acid (TCA) (Sigma Aldrich), for 1 h in 4°C, followed by three washings with water. The plate was then dried, and the cells were dyed with 100 µL/well of 0.057% SRB (Sigma Aldrich) in 1% acetic acid for 30 min in room temperature. The plate was washed with 1% acetic acid five times, dried, and 200 µL/well of 10 mM unbuffered tris-base solution were added to dissolve the bound SRB. The plate was shaken for 1 h and then read on SpectraMax at 560 nm. Results were normalized to values for non-treated cells.
Statistical analysis
All values were presented as mean ± SEM. Comparisons between multiple groups was assessed by one-way ANOVA with Tukey’s test. Comparisons between two groups were assessed by unpaired t-test, two-tailed. P values equal to or less than 0.05 were considered significant. Data represented as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
Results
CDDO-EA inhibits LPS-induced cytokine and chemokine production in macrophages
To examine the anti-inflammatory properties of CDDO-EA, we first determined whether CDDO-EA blocked cytokine and chemokine production in macrophages. We tested for the production of TNF-α because of the critical role this cytokine plays in the development of insulin resistance and T2D. 18 As seen in Figure 1, in RAW 264.7 mouse macrophages, LPS induced a dramatic rise in production of TNF-α. The rise in TNF-α was substantially blocked by pre-treatment with CDDO-EA. To further examine the properties by CDDO-EA in blocking expression of pro- and anti-inflammatory cytokines and chemokines in macrophages, THP-1 human macrophages were exposed to CDDO-EA prior to LPS treatment. Cytokines and chemokines were measured by a bio-plex assay. Table 1 and Figure 2 show that CDDO-EA primarily inhibited LPS-induced IL-1β, IL-6 and MCP-1 production. These findings are consistent with other reports showing that CDDO derivatives inhibit gene expression and secretion of pro-inflammatory mediators in immune cells.19–21
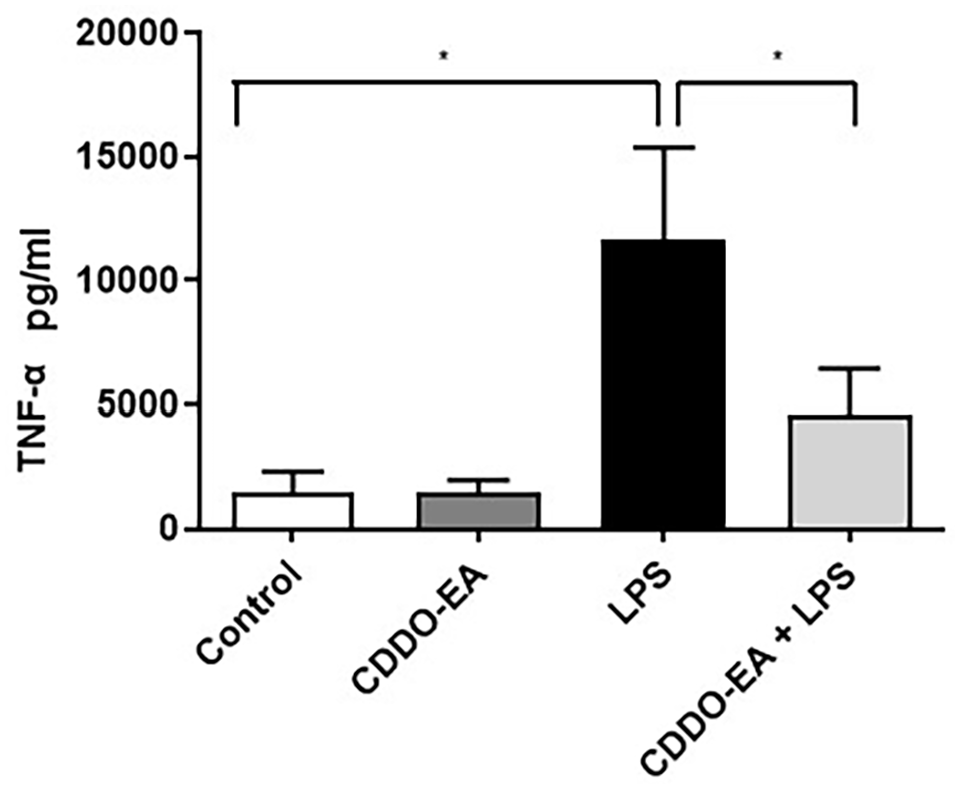
CDDO-EA inhibits LPS-induced cytokine secretion in macrophages. RAW264.7 mouse macrophages were pre-treated with 500 nM CDDO-EA for 1 h then exposed to 100 ng/mL LPS for 6 h. TNF-α levels were measured and quantified by ELISA. Three independent experiments were run in duplicate or triplicate. Data represented as mean ± SEM. *P < 0.05 (one-way ANOVA).
Human cytokine/chemokine panel in THP-1 macrophages exposed to CDDO-EA and LPS.
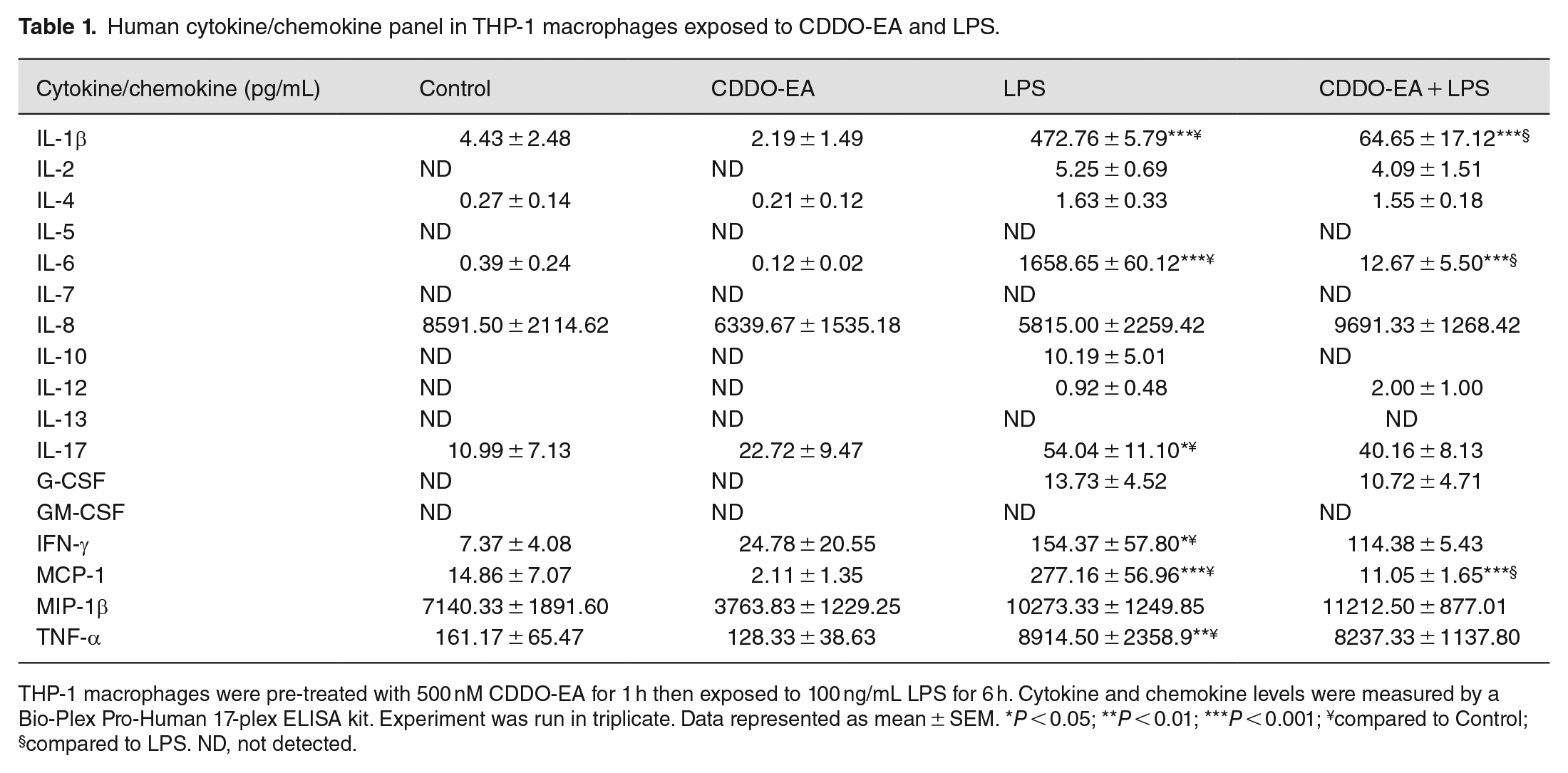
THP-1 macrophages were pre-treated with 500 nM CDDO-EA for 1 h then exposed to 100 ng/mL LPS for 6 h. Cytokine and chemokine levels were measured by a Bio-Plex Pro-Human 17-plex ELISA kit. Experiment was run in triplicate. Data represented as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ¥compared to Control; §compared to LPS. ND, not detected.
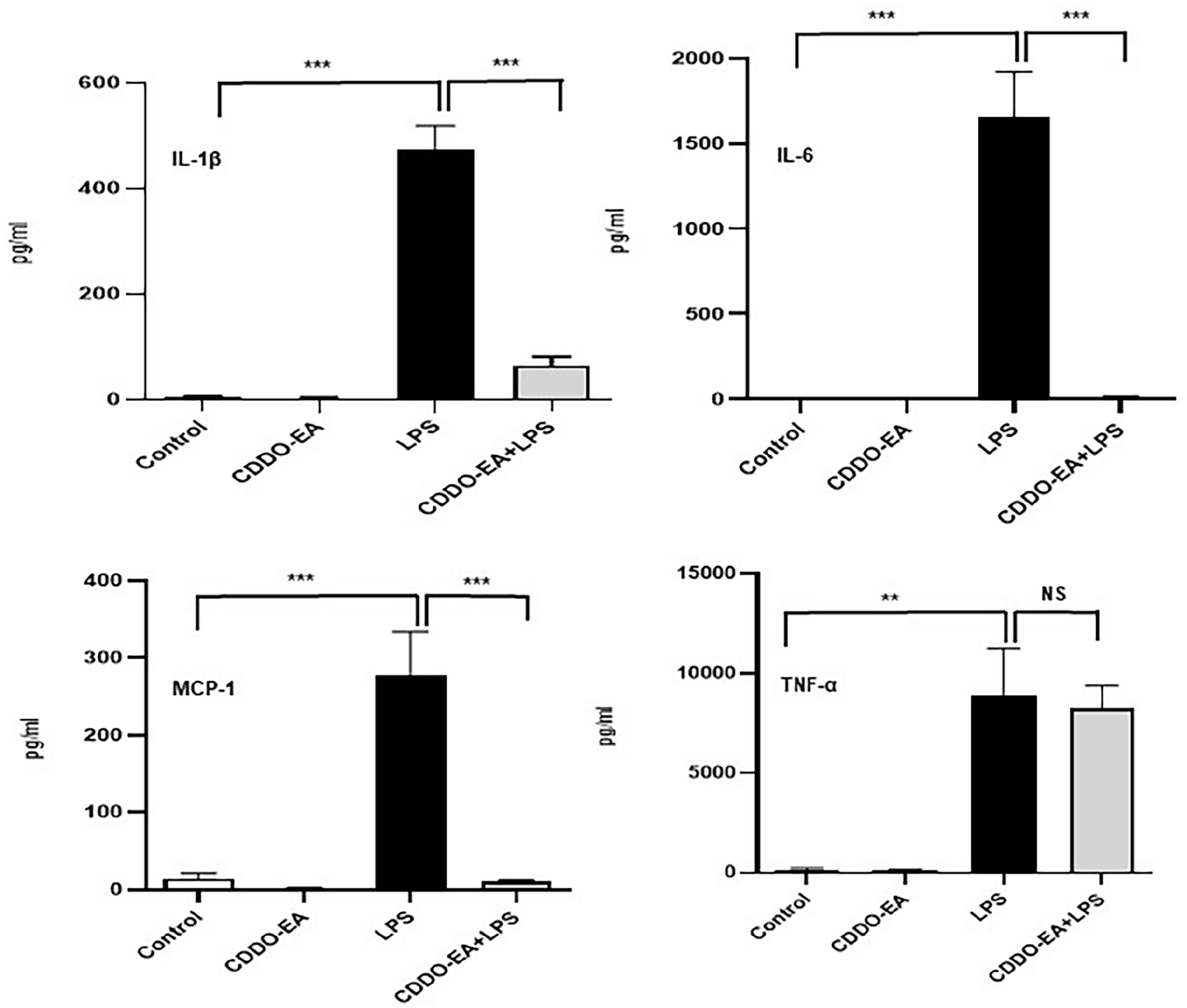
CDDO-EA blocks LPS-induced IL-1β, IL-6, and MCP-1 secretion in THP-1 macrophages. THP-1 macrophages were pre-treated with 500 nM CDDO-EA for 1 h then exposed to 100 ng/mL LPS for 6 h. Cytokine and chemokine levels were measured and quantified by a Bio-Plex Pro-Human 17-plex ELISA kit. Experiment was run in triplicate. Data represented as mean ± SEM. **P < 0.01; ***P < 0.001; (one-way ANOVA). NS: not significant.
CDDO-EA alleviates LPS-induced NF-κB and IκB phosphorylation in muscle cells
As shown in Figures 1 and 2, CDDO-EA can inhibit the production of pro-inflammatory cytokines and chemokines in macrophages. We next examined the anti-inflammatory properties of CDDO-EA on skeletal muscle. Specifically, we assessed the anti-inflammatory effect of CDDO-EA in inhibiting NF-κB activation in skeletal muscle. NF-κB is activated in skeletal muscle when exposed to LPS, resulting in the expression and production of pro-inflammatory cytokines. 22 CDDO-EA inhibited LPS-induced NF-κB phosphorylation in L6-GLUT4myc myotubes (Figure 3(A)). Upon activation of the inhibitor of nuclear factor kappa B kinase (IKK)/inhibitor of nuclear kappa B (IκB)/NF-κB pathway, the IKK complex phosphorylates IκB. 23 As shown in Figure 3(A), CDDO-EA also inhibited the LPS-induced phosphorylation of IκB-α in myotubes. To determine if 500 nM CDDO-EA effected cell viability, myotubes were treated with increasing doses of CDDO-EA for 6 h and 24 h. Myotubes treated with 6 h with 500 nM CDDO-EA showed 92% viability (Figure 3(B)). Cumulatively, these data show that CDDO-EA blocks LPS-induced activation of the IKK/IκB/NF-κB signaling pathway and that CDDO-EA at 500 nM does not induce cell death in skeletal muscle cells.
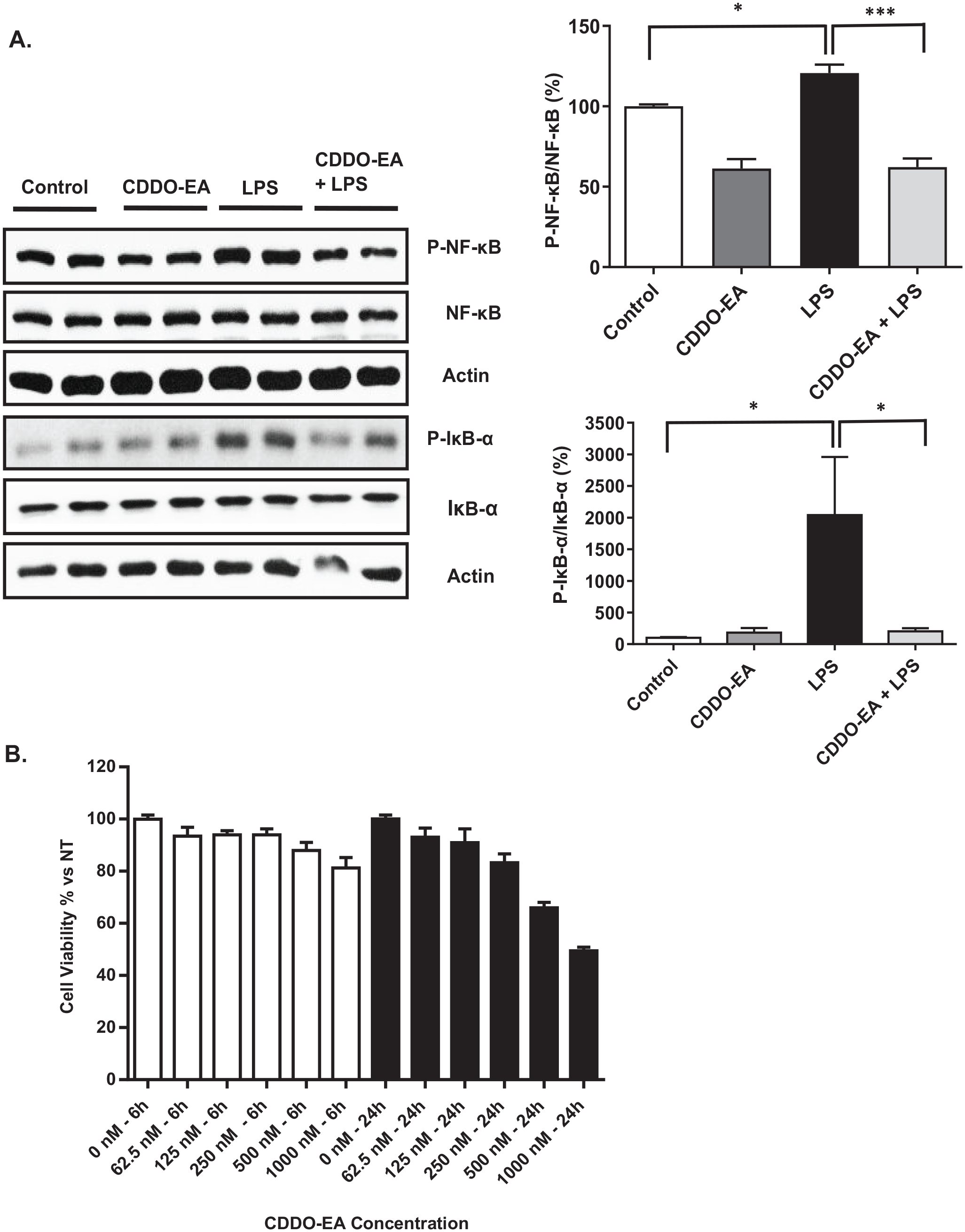
CDDO-EA inhibits LPS-induced NF-κB and IκB phosphorylation: (A) L6-GLUT4myc myotubes were pre-treated with 500 nM CDDO-EA for 1 h then exposed to 100 ng/mL LPS for 1 h. Myotubes were harvested for analysis by Western blot using antibodies against phosphorylated NF-κB p65 (Ser536), total NF-κB, phosphorylated IκB-α (Ser32), IκB-α total protein, and actin. NF-κB and IκB-α phosphorylation was normalized to NF-κB and IκB-α total protein, respectively, then expressed as percent over control sample. Four independent experiments were run in duplicate or triplicate. (B) L6-GLUT4myc myotubes were treated with doubling doses of CDDO-EA for 6 or 24 h. Cell viability was normalized to non-treated myotubes. Two independent experiments were run in triplicate. Data are represented as mean ± SEM. *P < 0.05; ***P < 0.001; (one-way ANOVA).
CDDO-EA blocks LPS-induced NF-κB nuclear translocation in muscle cells
NF-κB is bound to IκB in the cytoplasm. Upon activation of the IKK/NF-κB/IκB pathway, IκB is degraded, freeing NF-κB to translocate into the nucleus and activate the transcription of pro-inflammatory mediators. 23 To visualize the localization of NF-κB, immunofluorescence and confocal microscopy studies were performed in L6-GLUT4myc myotubes pre-treated with CDDO-EA for 1 h and then exposed to LPS for 1 h. The untreated muscle cells show a basal-level expression of p65 NF-κB in the nucleus (Figure 4(A)). LPS treatment for 1 h increases nuclear staining of p65 NF-κB, while pre-treatment with CDDO-EA reduces LPS-induced p65 NF-κB nuclear staining. We further examined nuclear translocation of p65 NF-κB by Western analysis of the nuclear and cytoplasmic fractions in CDDO-EA treated myotubes (Figure 4(B)). LPS exposure of 1 h increased nuclear localization of p65 NF-κB. However, treatment of myotubes with CDDO-EA for 1 h prior to LPS exposure decreased nuclear localization of p65 NF-κB.
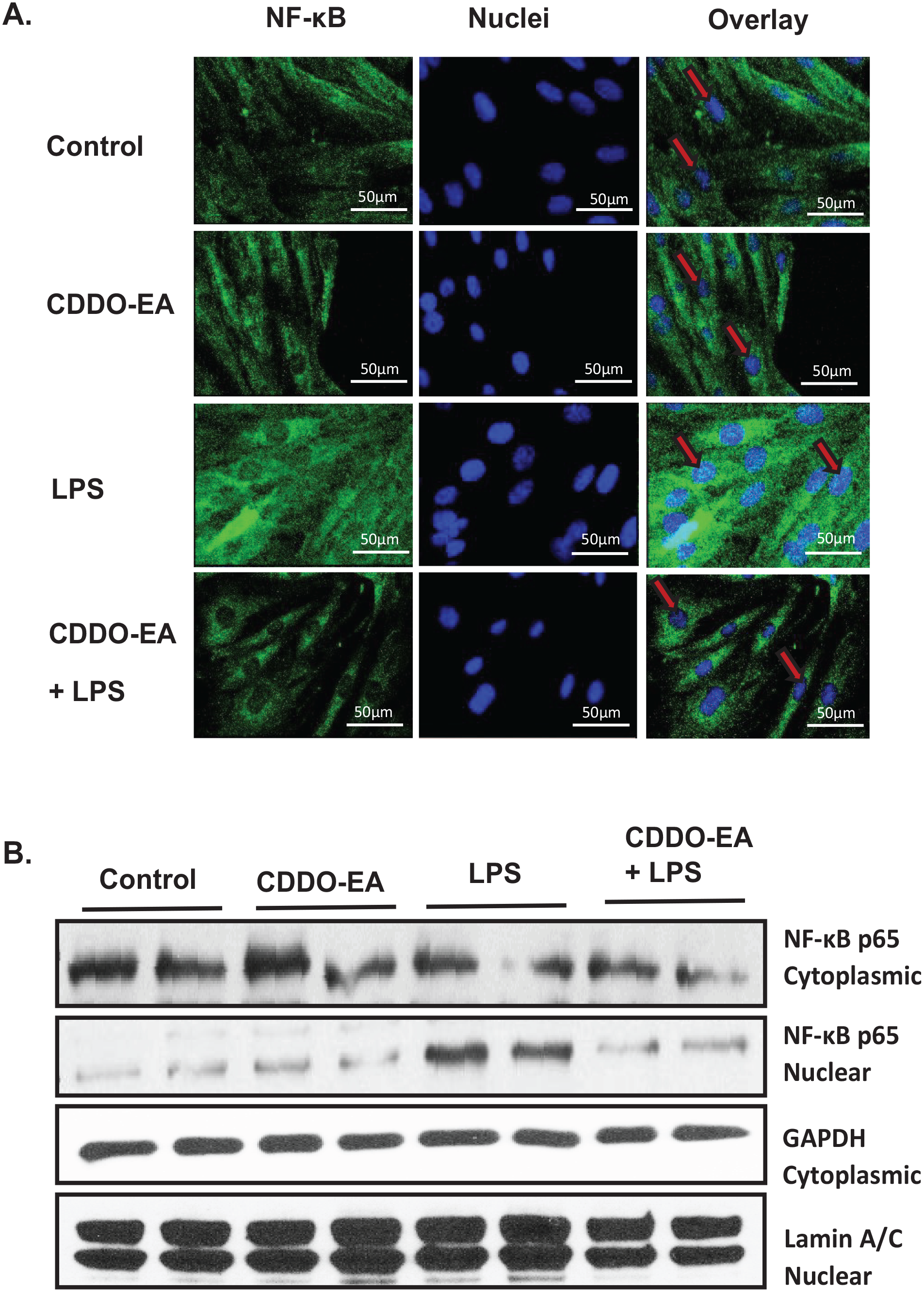
CDDO-EA affects NF-κB translocation. L6-GLUT4myc myotubes were pre-treated with 500 nM CDDO-EA for 1 h followed by 100 ng/mL LPS for 1 h. (A) For immunofluorescence, fixed, and permeabilized myotubes were stained using DAPI (nuclei, blue) and anti-NF-κB p65 primary antibody and a secondary antibody conjugated with Alexa Fluor 488. NF-κB was presented with green fluorescence in cytoplasm and nuclei (red arrows). Scale bar: 50 µm. Images are representative of two independent experiments. (B) Cytoplasmic and nuclear fractions were isolated and immunoblotted for p65 NF-κB and GAPDH and lamin to verify cytoplasmic and nuclear fractions, respectively. Data are representative of three independent experiments run in duplicate.
NF-κB activity and TNF-α and MCP-1 gene expression are inhibited by CDDO-EA in muscle cells exposed to LPS
To determine the effects of CDDO-EA on LPS-induced NF-κB transcriptional activity in L6-GLUT4myc myotubes, we used a luciferase reporter under the control of a promoter containing NF-κB transcription binding sites. LPS caused a 14.4-fold increase in NF-κB activity in myotubes at 6 h exposure (Figure 5(A)). Pre-treatment with CDDO-EA significantly inhibited the LPS-induced NF-κB activity. We next examined whether CDDO-EA blocked the mRNA expression of LPS-induced TNF-α and MCP-1, main pro-inflammatory mediators involved in the progression and development of T2D and transcribed by NF-κB.18,24 LPS stimulation of myotubes increased the expression of TNF-α and MCP-1 (Figure 5(B)). Pre-treatment of myotubes with CDDO-EA for 1 h reduced TNF-α and MCP-1 gene expression levels.
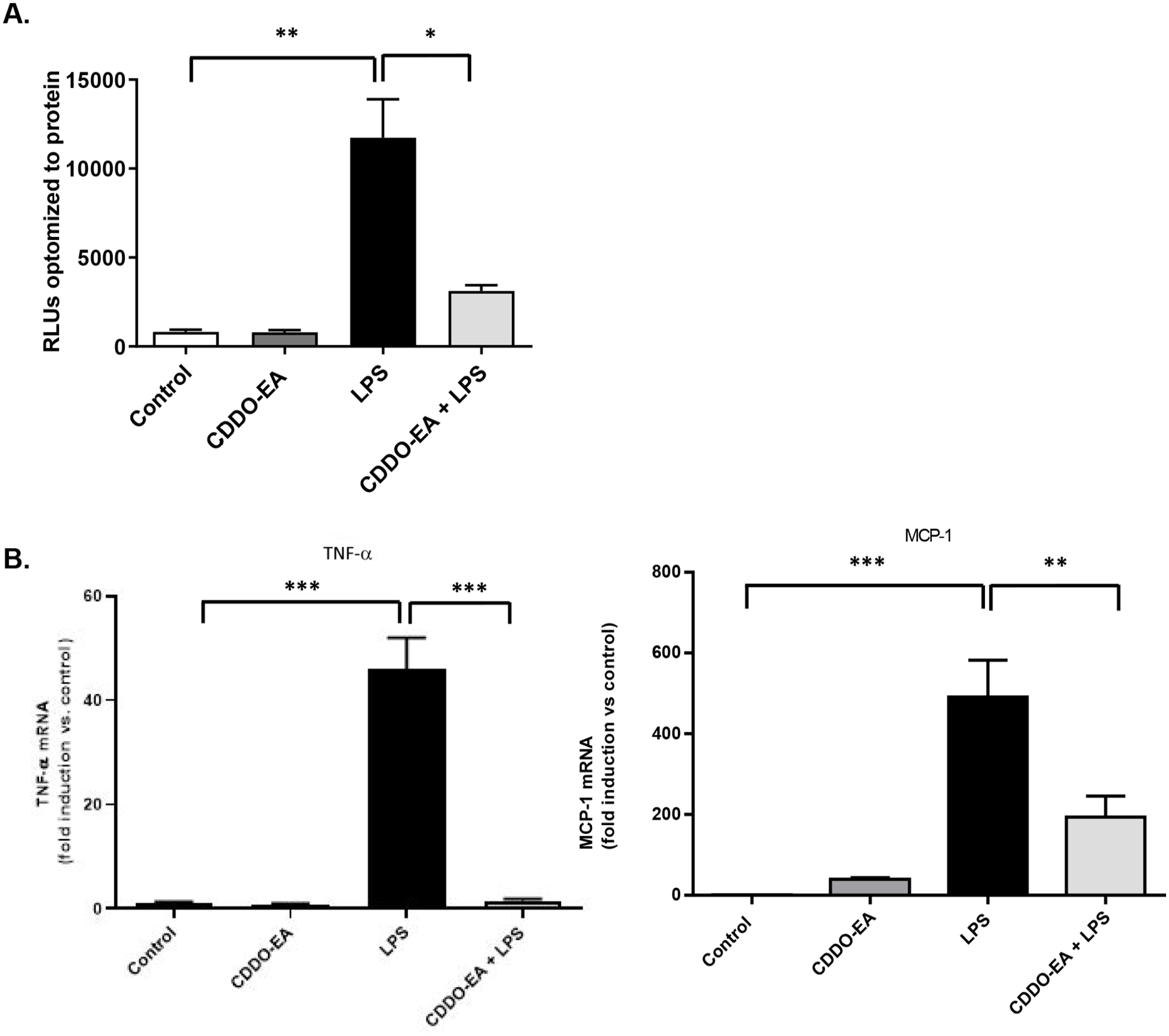
CDDO-EA blocks NF-κB transcriptional activity. (A) L6-GLUT4myc myotubes were transfected with an NF-κB luciferase reporter plasmid then treated with 500 nM CDDO-EA for 1 h followed by 100 ng/mL LPS for 6 h. Data indicate luciferase values normalized to total protein concentration. Experiment for each time point was run in duplicate. (B) GLUT4myc myotubes were exposed to 500 nM CDDO-EA for 1 h followed by 1 h exposure with 100 ng/mL LPS. mRNA expression levels of TNF-α and MCP-1 were measured by RT-PCR. Data are representative of two independent experiments run in triplicate (mean ± SEM). *P < 0.05; **P < 0.01 ***P < 0.001; (one-way ANOVA); RLU, relative light unit.
CDDO-EA induces insulin-independent GLUT4 translocation in muscle cells
We next examined the properties of CDDO-EA in regulating glucose metabolism in the skeletal muscle. We specifically determined whether CDDO-EA induced translocation of the glucose transporter, GLUT4. L6-GLUT4myc cells express c-myc epitope-tagged GLUT4 to distinguish from endogenous GLUT4 and express all the machinery for studying GLUT4 transport. 25 L6-GLUT4myc myoblasts differentiate normally from myoblasts to myotubes, and L6-GLUT4myc cells respond to insulin with a two-fold stimulation of glucose uptake and translocation of GLUT4myc to the cell surface. 25 In addition, L6-GLUT4myc myotubes express 100-fold more GLUT4myc than endogenous GLUT4 or GLUT1. 25 We performed immunofluorescence and confocal microscopy to visualize the localization of c-myc epitope-tagged GLUT4 in L6-GLUT4myc myotubes treated with CDDO-EA for 1 h. L6-GLUT4myc myotubes were also treated with insulin for 15 min as a positive control. As expected, insulin induced GLUT4myc translocation into the cell membrane (Figure 6(A)). Interestingly, CDDO-EA also induced GLUT4myc translocation, and LPS treatment for 1 h did not inhibit CDDO-EA-stimulated GLUT4myc translocation. Furthermore, because p38 has been reported to be involved in insulin-independent glucose metabolism,26–28 we examined if CDDO-EA activated phosphorylation of p38. Our findings show that CDDO-EA-induced p38 phosphorylation in skeletal muscle cells (Figure 6(B)).
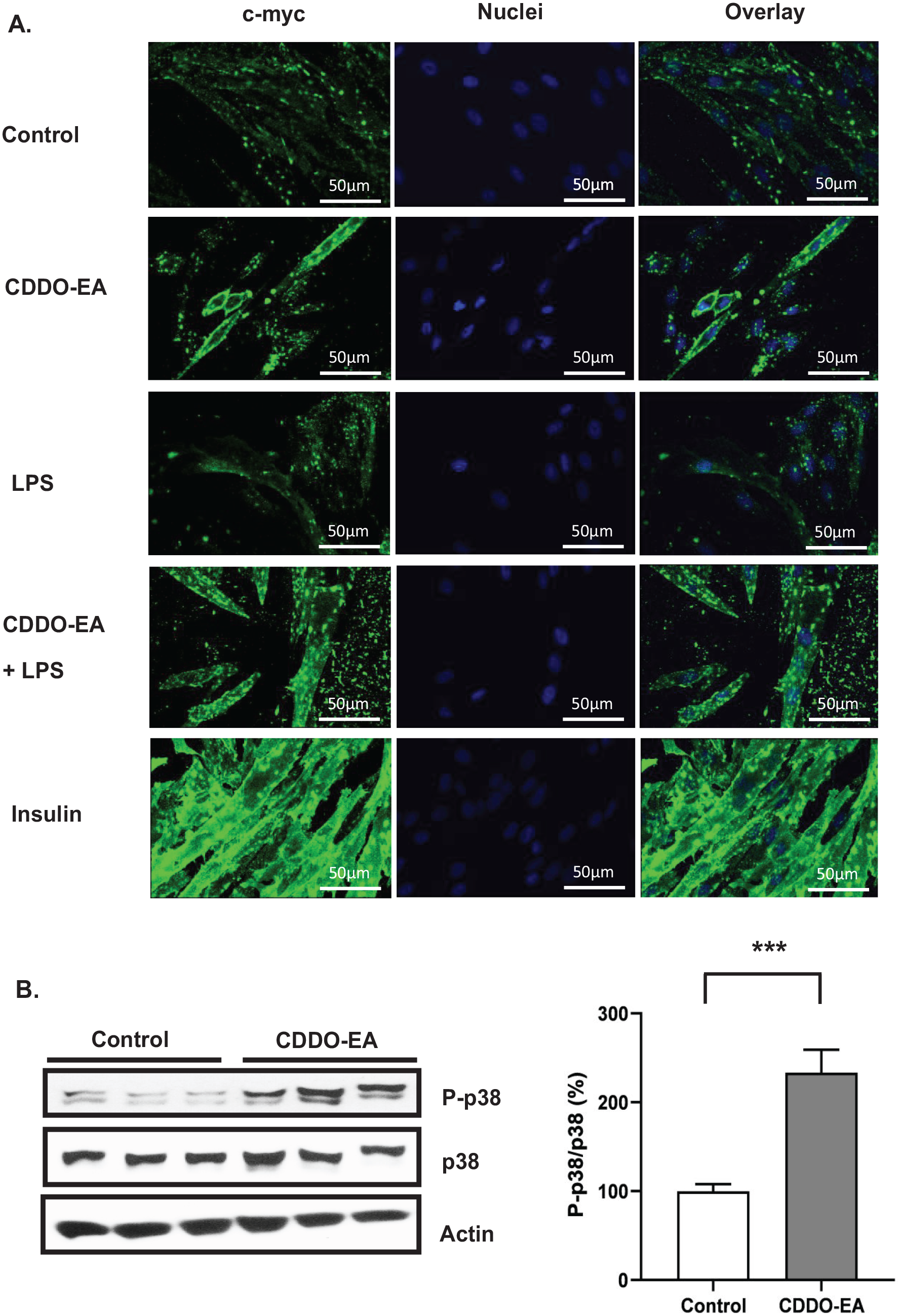
CDDO-EA activates GLUT4 translocation to cell membrane and p38 phosphorylation. L6-GLUT4myc myotubes were pre-treated with 500 nM CDDO-EA for 1 h followed by 100 ng/mL LPS for 1 h. 100 nM insulin treatment was for 15 min. (A) For immunofluorescence, myotubes were fixed without permeabilization and stained using DAPI (nuclei, blue) and anti-c-myc primary antibody (c-myc epitope-tagged GLUT4 distinguishes from endogenous GLUT4) and a secondary antibody conjugated with Alexa Fluor 488. c-myc was presented with green fluorescence. Scale bar: 50 µm. (B) Myotubes were harvested for analysis by Western blot using antibodies against phosphorylated p38, total p38, and actin. p38 phosphorylation was normalized to p38 total protein, then expressed as percent over control sample. Three independent experiments were run in triplicate. Data are represented as mean ± SEM. ***P < 0.001; (unpaired t-test, two-tailed).
Discussion
Because macrophages play a significant role in the initiation of chronic inflammation involved in the development of insulin resistance and T2D, we first wanted to determine if CDDO-EA suppressed cytokine and chemokine production in macrophages. This study demonstrated that CDDO-EA blocks TNF-α production in murine macrophages and prevents the production of IL-1-β, IL-6, and MCP-1 production in human macrophages when exposed to LPS. Similarly, studies demonstrated that pre-treatment with CDDO-Im attenuated the IL-6 and TNF-α gene expression in peripheral blood mononuclear cells (PBMCs) exposed to LPS. 29 PBMCs consist of a heterogeneous population of immune cells; therefore, Eitas and colleagues enriched monocytes from PBMCs and pre-treated these monocytes with CDDO-Me and then exposed them to LPS. 19 Our results were consistent with these findings in that CDDO-Me inhibited MCP-1 production; however, CDDO-Me did not reduce IL-6 production. A recent study showed that CDDO-Im decreased LPS-induced gene expression of MCP-1, IL-1-β, TNF-α, and IL-8 in macrophages differentiated from ML-1 monocytes. 21 The differences seen in the suppression of cytokines and chemokines could be due the use of macrophage cell lines compared to primary human immune cells and monocytes, the use of different derivatives of CDDO which can vary in their biological properties, and different anti-inflammatory mechanisms of action between cell types.
Published studies have mainly investigated the inhibition of NF-κB signaling by CDDOs in tumor cells because NF-κB is a survival factor in these cells therefore focusing on the pro-apoptotic properties of CDDOs.30,31 One study recently showed that CDDO-Im can inhibit LPS-induced NF-κB activity in RAW 264.7 cells. 21 However, the effects of CDDOs on inhibiting the activation of LPS-induced IKK/IκB/NF-κB signaling pathway in skeletal muscle is relatively unknown. NF-κB has long been implicated in the pathogenesis of insulin resistance and type 2 diabetes. 32 We have demonstrated that skeletal muscle of insulin-resistant individuals (obese and T2D) have increased activation of the NF-κB pathway and gene expression of IL-6, which is highly regulated by NF-κB.5,33 Furthermore, NF-κB signaling contributes to the establishment of a chronic and systemic inflammation by driving the transcription of pro-inflammatory cytokines and chemokines and contributing to the development of insulin resistance. 32 The present findings show that CDDO-EA inhibits the LPS-induced p65 NF-κB nuclear translocation, transcriptional activity, and gene expression of TNF-α and MCP-1 in skeletal muscle cells; thus, the mechanism of action for this inhibition is by suppressing the activation of the NF-κB signaling pathway. Our results also showed that CDDO-EA decreased cytoplasmic p65 NF-κB content, and this correlated with reduced NF-κB activity.
Although not much is known about the role of CDDOs in modulating GLUT4 translocation and glucose transport in skeletal muscle, one published report demonstrated that CDDO-Me treatment in mice fed an HFD increased insulin-stimulated glucose transport activity in skeletal muscle as measured by euglycemic-hyperinsulinemic clamp. 11 Our findings suggest that CDDO-EA induces insulin-independent GLUT4 translocation in skeletal muscle cells. Plus, our findings show that CDDO-EA activates p38. Published studies have demonstrated that p38 is implicated in insulin-independent GLUT4 translocation by skeletal muscle.26–28 For example, Rac1 GTPase, a known p38 effector,34,35 regulates exercise-stimulated GLUT4 translocation and glucose transport in mouse skeletal muscle in vivo. 26
Skeletal muscle is the major site of insulin-induced glucose uptake, so insulin resistance in skeletal muscle can dramatically affect systemic glucose metabolism. 1 It has been theorized that a cause for skeletal muscle insulin resistance may be due to systemic inflammation as a result of high circulating levels of LPS when a change in gut microflora occurs due to consumption of HFDs. 2 In addition, activation of skeletal muscle resident macrophages by LPS could affect skeletal muscle cells by the local release of cytokines and chemokines by these macrophages contributing to inflammation and development of skeletal muscle insulin resistance. 7 Our study provides a potential new role for CDDO-EA in protecting skeletal muscle from LPS-induced inflammation by blocking production of pro-inflammatory cytokines and chemokines in macrophages and inhibiting NF-κB activation, NF-κB nuclear translocation, and transcription of pro-inflammatory cytokines and chemokines in skeletal muscle cells. Also, our study suggests that CDDO-EA facilitates GLUT4 translocation and p38 activation as potential new mechanisms of CDDO-EA in regulating glucose metabolism.
Footnotes
Acknowledgements
The authors thank Dr. Amira Klip, Professor at the University of Toronto, for supplying the L6-GLUT4myc rat myoblast cell line and Dr. Thomas J. Slaga, Professor at the University of Texas Health Science Center at San Antonio, for providing CDDO-EA and for assistance with the initial design of experiments. The authors dedicate this work to the memory of their dear colleague and friend, Dr. Daniel Acevedo.
Authors’ Contributions
PF-MC and SMR designed experiments, analyzed and interpreted data, and wrote original draft; PF-MC and DA conducted experiments, collated and interpreted data; LJM interpreted data; PF-MC, SMR, and LJM participated in the review and editing of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by the National Institutes of Health [grant no. GM127272] and a UTRGV Institutional Seed Research Program Award.