
Abstract
The blood–brain barrier (BBB) is a critical physiochemical interface that regulates communication between the brain and blood. It is comprised of brain endothelial cells which regulate the BBB’s barrier and interface properties and is surrounded by supportive brain cell types including pericytes and astrocytes. Recent reports have suggested that the BBB undergoes dysfunction during normative aging and in disease. In this review, we consider the effect of cellular senescence, one of the nine hallmarks of aging, on the BBB. We first characterize known normative age–related changes at the BBB, and then evaluate changes in neurodegenerative diseases, with an emphasis on if/how cellular senescence is influencing these changes. We then discuss what insight has been gained from in vitro and in vivo studies of cellular senescence at the BBB. Finally, we evaluate mechanisms by which cellular senescence in peripheral pathologies can indirectly or directly affect BBB function.
Impact Statement
There have been significant advancements in understanding and characterizing the phenotype of cellular senescence over the last decade, and novel therapeutic approaches that eliminate senescent cells have advanced to clinical trials for a range of age-associated diseases, including those of the central nervous system (CNS). However, much remains to be learned about the predominant cell types that become senescent in the CNS and their functional effects. This review summarizes the known age-associated changes of the blood–brain barrier (BBB) induced by senescence, senescent changes in diseases affecting the CNS, and considers how senolytics may impact important BBB functions.
Introduction
Cellular senescence
Cellular senescence is a complex phenomenon first identified by Hayflick and Moorhead 1 in 1961. In their seminal publication, which has now been cited over 1800 times, they described isolated human fibroblasts having a “finite limit of cultivation.” That is, after about 50 subcultures, these cells “degenerated” and stopped proliferating. Further research determined that senescent cells accumulate with age and established cellular senescence as a hallmark of aging.2,3 Today, cellular senescence is generally defined by an irreversible arrest of the cell cycle with accompanying stereotyped phenotypic changes.
In the past 20 years, there has been significant insight into the phenotype of senescent cells; thorough phenotypic characterization is crucial as there is yet to be any universal and specific markers to determine whether a cell is senescent. Beyond irreversible cell-cycle arrest (regulated by p16INK4a and/or p53/p21),4,5 features of senescent cells include, but are not limited to, secretion of inflammatory proteins (e.g. pro-inflammatory cytokines and chemokines, angiogenic factors),6,7 high lysosomal activity (e.g. increased senescence-associated β-galactosidase, SA-β-gal), 21 morphological differences (e.g. cell blebbing), 8 and nuclear changes (e.g. downregulated lamin B1, Figure 1(A)). 9 Due to the heterogeneity of cellular senescence, the International Cell Senescence Association published their consensus on how to identify cellular senescence, outlining detailed cellular and molecular changes, and we refer the reader to these excellent analyses.10,11
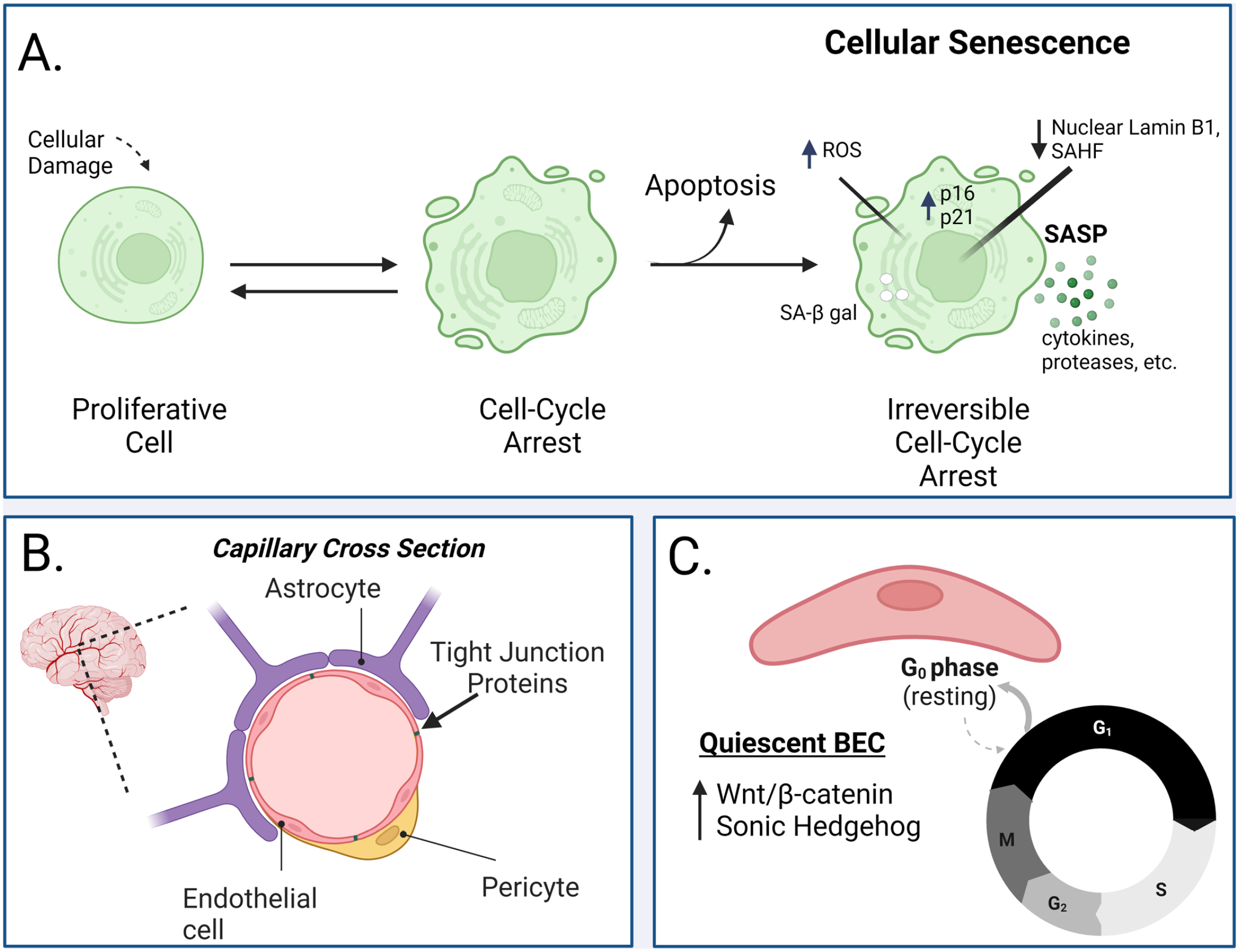
Cellular senescence and the BBB. (A) A variety of different stressors can induce cell-cycle arrest including telomere attrition, oxidative stress, DNA damage, and epigenetic stress. From here, a cell can mend itself via internal mechanisms and re-enter the cell cycle or, if the damage is overtly destructive, a cell can turn on apoptotic programs to prevent damaged machinery from impacting the health of the tissue. However, in some cases (the mechanisms by which are still under investigation), a cell can stay in cell-cycle arrest and, over an undetermined period of time (anywhere between 10 days to 6 weeks in vivo), enters a state of cellular senescence where it can no longer re-enter the cell cycle. Besides irreversible cell-cycle arrest, the phenotype of cellular senescence consists of (but is not limited to) increased p16 and p21/p53, decreased nuclear lamin B1, senescence-associated heterochromatin foci (SAHF), increased senescence-associated β-galactosidase, increased reactive oxygen species, and a senescence-associated secretory phenotype (SASP) which is a pattern of secreting cytokines and proteases. (B) The BBB primarily consists of brain endothelial cells (BECs, pink) which are sealed together by tight junction proteins. BECs are embedded in a basement membrane and are physically surrounded by pericytes (yellow) and astrocytes (purple) in the brain which support BBB induction and maintenance as important components of the neurovascular unit (NVU). The schematic is of a capillary cross-section in the brain. (C) BECs predominantly exist in a quiescent state (G0 resting phase of the cell cycle). This process is regulated by Wnt/β-catenin as well as sonic hedgehog signaling. Quiescent BECs can re-enter the cell cycle with appropriate stimuli. This figure was made with BioRender. BBB: blood–brain barrier.
Cellular senescence does have beneficial features in that it prevents proliferation of damaged cells and triggers their breakdown by the immune system. However, the onset and prolonged duration of cellular senescence can lead to dysfunctional stem cells and widespread inflammation attributed to the senescence-associated secretory phenotype (SASP). This chronic senescent phenotype has been identified as a contributor to the aging brain and age-related brain diseases including Alzheimer’s disease (AD) and frontotemporal dementia.12,13 In 2015, the first senolytic therapeutic strategy, a combination of dasatinib and quercetin, was reported, 14 which boosted enthusiasm and heightened interest in the geroscience community. At the time of submission of this review, there are 16 active clinical trials that look at the effect of senolytics in a variety of age-associated diseases including chronic kidney disease, osteoarthritis, and AD.
The discovery of senolytics has strengthened the research into cellular senescence and its influence in disease. Recent studies have explored evidence for senescence in brain cells including astrocytes, 15 microglia, 16 and neurons. 17 In addition, there is significant evidence for endothelial cell senescence in age-related peripheral diseases as well as vascular-associated disease.18,19 Central nervous system (CNS) vascular dysfunction, including impairment of the microvascular blood–brain barrier (BBB), can be a major contributor to age-associated CNS diseases. However, relatively little is known about the extent that cellular senescence contributes to age- and disease-associated changes of the BBB. In this review, we will discuss the current literature that has elucidated the relations of cellular senescence to BBB dysfunction.
The vascular BBB
The vascular BBB is an essential brain interface primarily comprised of brain endothelial cells (BECs) that protect the brain from exposure to potentially harmful substances in the circulation and regulate the transport of essential nutrients and signaling molecules between brain and blood compartments (Figure 1(B)). The BBB’s interface and barrier properties are conferred, in part, by unique BEC features that include low levels of pinocytosis, tight junction proteins that limit paracellular leakage, and transporters that regulate which substances can enter or leave the brain. 20 BECs are surrounded by supportive brain cell types including astrocytes and pericytes which make up the neurovascular unit (NVU). Many excellent reviews highlight the multifaceted features of the BBB in health and development, as well as in aging and disease.20–22 Recently, multiple modes of BBB dysfunction have been associated with aging, including increased leakiness, impaired transport of molecules such as glucose, 23 amyloid-beta peptide,24,25 and xenobiotics.26,27 It is theorized that these changes at the BBB may be drivers of cognitive dysfunction in aging and may be further impaired in neurodegenerative diseases like AD.28–34 Several reports have sought to understand how epigenetic stress and mitochondrial dysfunction act; two of the nine “hallmarks of aging” contribute to BBB dysfunction.35,36 In this review, we focus on a third: cellular senescence.
Quiescent resting state of BECs
As background, it is important to distinguish between quiescence and senescence. In contrast to senescence, which is generally irreversible, 10 quiescence is a generally reversible state of cell-cycle arrest that is essential for certain types of cells with strict proliferative guidelines such as lymphocytes and stem cells. 37 Quiescent cells can share some features of senescent cells, such as increased lysosome content/SA-β-gal activity, although the molecular underpinnings can differ. 38 Notably, the BBB, and in particular BECs, primarily exist in a quiescent state (Figure 1(C)). 39 Quiescence is regulated by nutritive and growth factors and/or contact inhibition, and maintains cells in a stable state where they are non-proliferative, yet able to re-enter the cell cycle with appropriate stimuli. In the mature, healthy BBB, the quiescent endothelium exhibits very little proliferation/migration, vascular leakage, and expression of leukocyte adhesion molecules. 39 Wnt/β-catenin and sonic hedgehog signaling regulate vascular quiescence and are crucial to the integrity of the BBB.39,40 Conversely, cellular senescence can occur in response to a variety of cell stressors (including DNA damage, oncogenic mutations, telomere attrition, and oxidative stress).41–44 This review will specifically examine cellular senescence and its involvement at the BBB.
Aging
First, we will review changes at the aging BBB such as permeability, morphology, and transport kinetics. We include core features of the BBB including the basement membranes (BMs) and glycocalyx, as well as consider the effect of advanced age and relative systemic changes such as blood pressure and pulse flow.
Normative age–related changes at the vascular BBB
The vascular BBB has long been known to undergo morphological and functional changes with normative aging. 22 Studies have shown that brain capillaries have a decreased number of BECs that become more elongated with age. 45 Furthermore, there is an overall decrease in capillary diameter, particularly BECs in white matter, due to loss of cytoplasm. 46 This thinning is thought to be related to a loss of pericytes. 46 Mitochondrial number and gap junctions in BECs do not vary, which proposes that some facets of the BBB remain intact with healthy aging. More recent studies have found decreased tight junction expression 47 and evidence for a shift toward increased transcytosis 48 at the BBB with age, indicative of changes in BBB structure and function.
Whereas morphological studies found little or no evidence for BBB disruption, immunohistochemical studies have been mixed. Cuffing studies of albumin, fibrinogen, or immunoglobulin G (IgG) occurs in young as well as aged brains, with a variance too large to allow conclusions. 49 Cerebrospinal fluid (CSF)/serum ratios of albumin do increase with healthy aging, 50 but may reflect decreased CSF reabsorption rather than or in addition to BBB disruption. 51
Imaging studies using dynamic contrast-enhanced magnetic resonance imaging (MRI) have found that in healthy humans, there is a modest increase in permeability of a low-molecular weight gadolinium-based tracer in the hippocampus that occurs with age. 32 This compares to early tracer studies, which found little evidence for disruption of the BBB to sucrose, 52 albumin, 53 or horseradish peroxidase. 54 Another imaging study found increased leakage in the BBB of white matter, gray matter, and the hippocampus, where the white and gray matter leakage correlated with cognitive decline. 55 It should be noted that the degree of increased leakage that occurs with healthy aging is small and highly variable among individuals. In fact, one study using dynamic contrast-enhanced MRI found that a 90+ individual had BBB integrity matching the 20-year-old group. 55 In retrospect, the small increase and large variation explains why it has been so difficult for morphometric and immunohistochemical studies to detect disruption. Therefore, while there is a trend toward increased BBB disruption with age, this is not a universal feature of aging.
Aging is associated with changes in the transport kinetics of several substances. Peptide transport system-1 56 and the brain-to-blood transport of amyloid-beta peptide 57 decrease with aging. A reduced transport of interleukin-1 beta across the BBB has been postulated to underlie the diminished fever seen with aging. 58 Other transporters that are altered with normative aging are the large neutral amino acid transporter, the efflux transporter P-glycoprotein, and the transporters for choline, triiodothyronine, tumor necrosis factor-alpha, and glucose. 22 It is unclear the degree to which the decrease in BBB transport systems arises from a defect in BBB function versus an adaptive response of the BBB to a decreased demand for transporter substrates by the brain. The composition of the lipid membrane dictates the degree to which small, lipid soluble substances can cross the BBB by way of the non-saturable mechanism of transcellular diffusion. Fluorescence polarization studies suggest that membrane composition of BECs changes little with aging. 59
As the above illustrates, the vascular BBB undergoes structural and functional changes with normative aging. This raises the question as to whether the age-related changes in BBB are accompanied by cellular senescence of BECs of which we detail in a later section.
Normative age–related changes at the BBB extracellular matrix
Senescent cells and their associated secretory phenotype promote dysfunctional tissue turnover, which results in depletion of extracellular matrix (ECM) in some tissues and abnormal matrix deposition in others.60,61 For the brain in general, and the microvascular ECM in particular, there is an overall increase in degradative processes that is often attributed to matrix metalloproteases-2 and -9 (MMPs).62,63 The higher protease activity found in aged tissues is usually not accompanied by similar increases in inhibitors of activity, such as the tissue inhibitors of MMPs.64,65 The resulting milieu can affect the ECM on both the luminal and abluminal side of the BBB.
The luminal ECM is known as the glycocalyx, a dynamic thin gel-like layer present along the endothelium throughout the macro- and micro-vasculature. The glycocalyx is comprised primarily of heparan sulfate and chondroitin sulfate proteoglycans, non-sulfated glycosaminoglycans, and their associated binding proteins, which are constantly modified by interactions with the circulation.66,67 Content and synthesis of hyaluronic acid, a component of the glycocalyx, increases with aging. 68 Furthermore, aging is associated with a thinner glycocalyx and lower barrier function in the peripheral vasculature. 69 Thinning of the glycocalyx is multifactorial and reflects increases in protease activity and oxidative processes; at the same time, there are decreases in protective factors such as heparan sulfates and sirtuins. 70
On the abluminal side of the microvasculature is the BM, a thin and intricate layer of ECM that supports the BECs and invests the pericytes. The BM is composed of an intercalating network of laminin, collagen IV, and fibronectin interspersed with proteoglycans and glycoproteins. 71 In contrast to the dynamic turnover of the glycocalyx, the BM is relatively stable with sheets of collagen IV fibers and laminin chains that are interwoven with a repository of deposition products and post-translational modifications. The BM can thicken during normative aging, a process that is accelerated by disease processes.72,73 For example, advanced glycation end products are non-enzymatic, post-translational modifications of proteins such as type IV collagen that are increased in aging, hypertension, diabetes, and inflammatory processes that increase carbonyl stress. Several studies have demonstrated that these alterations interfere with BBB function.74,75
Advanced age and relevant systemic age-related changes
During normative aging, there are increases in systolic blood pressure and pulse pressure, with changes in diastolic blood pressure being more variable. As a result, there is an increase in the prevalence of the syndrome of isolated systolic hypertension in older adults. 76 The effect of widened pulse pressure on brain microvasculature is typically implied rather than directly studied, but there is increasing evidence that higher intravascular stress has a detrimental effect on BBB function and negatively impacts glymphatic clearance.77,78 The beneficial effect of treating hypertension on cognitive outcomes may reflect a dampening of subsequent damage to the brain microvasculature that could include stabilization of BBB ECM.79,80 Studies that administered chronic senolytics to mice suggest benefits include reducing calcifications in the vascular ECM and improving nitric oxide bioavailability to the general vasculature. 81
The off-target effects of the numerous medications that are taken by an aging population could affect cellular senescence. The range of medications that potentially influence the microvasculature is beyond the scope of this review, but certain classes of commonly used medications likely improve BBB function and mitigate senescence changes. Drugs affecting the latter would include medications that lower systolic blood pressure or glucose, such as angiotensin inhibitors, selective sodium-glucose cotransporter 2 inhibitors, and glucagon-like peptide-1 receptor agonists.82–84 The increasing use of biologics that can alter the entire immune system might dampen inflammation seen with aging (known as inflammaging) and subsequent senescence, but could also detrimentally affect compensatory processes that ward off infections. 85 Future studies will establish how senolytics that mitigate the senescent changes in the BBB influence the effect of medications on the aged brain.70,86
Brain diseases
Neurodegenerative diseases
Neurodegenerative diseases are associated with both BBB dysfunctions and with aging.87,88 This raises the possibilities that accelerated senescence of BECs could be the driver for BBB dysfunction in neurodegenerative diseases or that neurodegenerative diseases can accelerate the aging of the BBB with the accumulation of senescent BECs. BBB dysfunctions, cellular senescence, and neurodegenerative diseases all have common factors in their pathology including the mechanisms of oxidative stress, mitochondrial dysfunction, and inflammation. Therefore, one would predict that many neurodegenerative diseases or age-associated diseases with altered CNS function are associated with both BBB dysfunctions and cellular senescence. It is clear, given that senescent cells can induce other cells to become senescent in a paracrine-like fashion, that cellular senescence in other cells of the NVU is important in BBB dysfunction. It is also supported that BBB dysfunction can lead to cellular senescence in other, adjacent cells. For example, Preininger and Kaufer have reviewed how an aging, leaky BBB can induce TGFβ signaling, leading to senescence in astrocytes. 89 However, there are relatively few studies examining BEC senescence in neurodegenerative diseases to date. Brain microvessels from subjects with higher levels of tauopathy have upregulation of senescence-related genes. 90 This suggests that tau pathology can contribute to cellular senescence in BECs. More studies are necessary to study effects of other neurodegenerative proteinopathies like Parkinson’s disease (alpha synuclein) and AD (amyloid beta).
Contribution of apolipoprotein E, a common genetic risk factor for AD
Apolipoprotein E (apoE) is a lipoprotein component that helps shuttle lipids and cholesterol around the body. 91 In humans, apoE is primarily produced by the liver in the periphery, and by astrocytes and microglia in the CNS. Because plasma apoE does not enter the brain, it is possible that peripheral and CNS sources of apoE differentially influence disease processes. There are three isoforms of human apoE: E2, E3, and E4. In AD, apoE4 is the greatest known genetic risk factor and thus heavily studied in neurodegenerative contexts. 92 To date, there have been very few studies investigating the impact of apoE genotype on cellular senescence, particularly at the BBB. In vitro studies have shown quercetin, one component of the senolytic cocktail of dasatinib plus quercetin, can prevent the enhanced redox state of apoE4-treated neuroblastoma cells (SK-N-SH cells) potentially by modifying the structure of apoE4. 93 As oxidative stress can lead to senescence, preventing this E4 modification could slow this process. Other accumulating evidence suggests that apoE4 accelerates cellular senescence of microglia. 94 Cellular apoE levels can accelerate senescence by destabilizing heterochromatin, while loss of apoE resists senescence; 95 therefore, a change in the cellular apoE level could trigger senescence of a given cell, but these studies remain to be performed in BECs. However, while apoE genotype can regulate apoE protein level, apoE4 produces less apoE protein compared to apoE3 or apoE2, 96 suggesting an apoE4 genotype is not likely to lead to senescence via heterochromatin destabilization.
Importantly, human clinical trials are just beginning to investigate the impact of senolytics in AD and whether there are apoE genotype effects of cellular senescence. There is currently a National Institutes of Health (NIH) grant committed to investigating the impact of the apoE genotype on senescence in AD and vascular cognitive impairment and dementia. 97 In a preclinical study, mice overexpressing AD genes (5XFAD) crossed with apoE3/E4 transgenic mice (E3FAD, E4FAD) were fed rapamycin for 16 weeks, beginning before the onset of amyloid-beta peptide deposition, which is a distinct pathological feature in AD. 98 E4FAD mice had decreased levels of p-glycoprotein (Pgp), an efflux transporter at the BBB, and increased accumulation of amyloid-beta peptide. When fed rapamycin, a strategy that reduces the SASP phenotype (also known as a senomorphic),99,100 Pgp protein and amyloid-beta peptide levels were restored to the E3FAD level. Cerebral blood flow was also improved in E4FAD rapamycin treated animals indicating a link between cellular senescence and amyloid beta transport at the BBB. Since a senescent phenotype was not assessed in this study, there is no way to identify whether the improvements from rapamycin were due to an impact on senescence or related to its inflammation-suppression role.
apoE genotype may also influence the biological activities of senolytics. While senolytic combinations have not yet been tested, an apoE genotype effect was shown to occur in response to dietary quercetin. Following six weeks of quercetin intake, apoE3 mice had a significant reduction in blood tumor necrosis factor α (TNF-α) levels (44%) while apoE4 mice did not have a significant reduction. 101 TNF-α can directly impact the BBB 102 or can cross the BBB to act within the CNS 103 and can induce senescence in many cell types, including endothelial cells.104,105 The limited data on apoE genotype in BBB senescence clearly warrant further investigations based on the intersecting signaling pathways involved and data reported in peripheral cells.
In vitro/in vivo studies inducing cellular senescence at the BBB
As previously described, cellular senescence is one of the nine hallmarks of aging, 2 and BBB dysfunction can also occur with normative aging. 106 Both processes may be exacerbated by underlying diseases (which will be discussed). However, the extent to which brain microvascular senescence contributes to BBB dysfunction is less clear. This section reviews our current understanding of the relation of brain microvascular senescence to BBB dysfunction based on findings in cell models, rodents, and from single-cell RNA sequencing studies of human BECs.
Brain microvascular/endothelial senescence and BBB dysfunction in rodents
As discussed in previous sections, BBB dysfunction appears to be a feature of normative aging that is exacerbated in neurodegenerative disease and with associated risk factors. Aged mice have been reported to have a leaky BBB,34,107 which can be mediated in part through the decreased expression of endothelial sirtuin-1, 34 an inhibitor of endothelial cell senescence.108,109 Recent analysis of single-cell RNAseq data sets from young versus aged mouse brains found that the percentage of senescent BECs increased from 5.23% to 10.06%. Notably, the method used to calculate this increase relied on enrichment scores for a panel of senescent gene markers, taking into account both the expression levels and numbers of markers expressed in each cell. 110 The authors of this study reported unpublished findings that a similar increase in BEC senescence was found in the p16-3MR senescence reporter mice. Senescence-associated BBB dysfunction has also been demonstrated in rodent models of accelerated senescence. BubR1 hypomorphic mice (BubR1H/H) have a mutation in the BubR1 gene, resulting in reduced levels of the protein. BubR1 is a mitotic checkpoint activator protein that regulates proper chromosomal segregation and attachment to the mitotic spindle. 111 This protein is reduced with aging, and BubR1H/H mice are prone to aneuploidy and develop a progeroid phenotype which includes growth retardation, short lifespan, impaired wound healing, cardiac arrhythmias, and arterial wall stiffening.111,112 Cellular senescence is widespread in many tissues in this model. The introduction of the INK-ATTAC transgene, which expresses a drug-activatable apoptosis-inducing caspase-8 and fluorescent reporter driven by the promoter for p16Ink4a, in BubR1H/H mice results in selective elimination of senescent cells with transgenic caspase-8 activating AP20187 treatment, and improvement of aspects of the aging phenotype. 112 Increased SA-β-gal staining and p16Ink4a mRNA were also noted in cultured BECs and pericytes of BubR1H/H mice versus wild-type (WT) controls. 113 In vivo, it was shown that five-month-old BubR1H/H mice have increased BBB disruption, as measured by greater leakage of the tracers Evans blue and sodium fluorescein into the cerebral cortex in comparison to the WT controls. Reduced levels of tight junction proteins claudin-5, ZO-1, and occludin were also measured in the cerebral cortex of BubR1H/H mice versus WT controls 113 Moreover, a recent study that the stabilization of G-quadruplexes upregulates cysteine rich angiogenic inducer 61 (CCN1) and induces cellular senescence in BECs. 114 CCN1 is a matricellular protein secreted by BECs which binds to heparin sulfate proteoglycans, activating DNA damage factors, inducing expression of MMPs that are part of the SASP, and triggering cellular senescence. There was an age-associated increase in CCN1 from cerebral microvessels isolated from young (4 months) versus old (12 months) mice that was concomitant with increased senescence features. Together, these data suggest an association of brain microvascular senescence with BBB disruption; however, more mechanistic studies are needed for definitive evidence that cellular senescence in BECs or other cells of the NVU is driving the BBB disruption phenotype.
Induction of senescence in BBB cells in vitro
In vitro models of the BBB/primary cultures of BECs and other cells of the NVU have also been used to study aspects of cellular senescence. As initially described by Hayflick and Moorehead 1 in the 1960s, replicative senescence occurs when cells have surpassed their proliferative lifespan, which can be achieved by serial passaging. Stamatovic et al. 34 used two immortalized models of BECs to study effects of passage and hydrogen peroxide-induced senescence on BBB permeability. They found that both models of senescence were associated with increased levels of senescence markers and increased BBB permeability. Moreover, Stamatovic et al. found that primary mouse BECs underwent nearly complete senescence (as defined by SA-β-gal staining, upregulation of p21, DNA damage marked by γH2A.X, and heterochromatin foci) after six complete population doublings. Senescent mouse BECs showed increased BBB leakage as evidenced by reduced transendothelial electrical resistance, and increased permeability to albumin in comparison to non-senescent/low passage mouse BECs.115–117
Senescence can also be induced in vitro through physical or chemical stressors, such as ionizing radiation, chemotherapeutics, or oxidative stress and specific protocols have been described to do so.115–117 Importantly, senescence is a slow process, taking about three to seven days to develop in response to stressors. 118 Ionizing radiation of rat BECs elicited DNA damage, which resulted in greater cell death, while repair of DNA damage was slower than in neurons, astrocytes, and microglia. While ionizing radiation caused apoptosis in about 10% of cells, about 30% of surviving cells showed hallmarks of senescence that included SA-β-gal staining, increased p16Ink4a and p53, an SASP, and reduced proliferative capacity. Oxidative stress was also elevated. 119 Phenotypic characterization of cultured BECs and pericytes from young (5–8 weeks old) versus middle-aged (40–50 weeks old) mice showed that both cell types exhibited increases in SA-β-gal staining and reduced proliferation with age. Interestingly, whereas endothelial cells from older mice had significant elevations in p21 expression but not p16INK4a, pericytes had significant elevations in p16INK4a but not p21. 113 In a triculture model of primary BECs, astrocytes, and pericytes, it was shown that BECs from BubR1H/H mice were more leaky as measured by TEER and leakage to sodium fluorescein and albumin, and had abnormal expression and mislocalization of the tight junction proteins claudin-5, ZO-1, and occludin. 113 BECs and pericytes from BubR1H/H mice had increased SA-β-gal staining and increased expression of p16INK4a versus WT mice. 113 Similar to results in vivo, these studies support that BBB dysfunction is associated with cellular senescence, and that endothelial cells with a senescent phenotype form a less robust BBB in vitro.
Mechanisms by which cellular senescence could influence BBB functions
In the previous section, an overview was provided on current knowledge of how senescent changes particularly in BECs are associated with BBB dysfunction. However, theoretically senescent cells in the brain or even in the periphery could contribute to aspects of BBB dysfunction through their direct and indirect interactions with BEC (Figure 2). Cells of the NVU such as astrocytes and pericytes have established roles in supporting the development and maintenance of the BBB.120,121 Currently, there is minimal evidence informing to what extent these cell types become senescent in vivo; analysis of an existing single-cell RNAseq data set did not detect senescent pericytes, smooth muscle cells, or astrocytes in aged mice. 122 Astrocyte senescence has been observed in a number of in vitro models of disease-associated stimuli, and it is known that astrocytes in vivo adopt a more pro-inflammatory phenotype with aging and in CNS diseases. 15 In vivo, p16INK4a positive astrocytes were found to be increased in the frontal cortex of human brains with aging and AD. 63 Microglia can also have physical associations with the BBB in health and disease, resulting in beneficial or harmful effects on BBB functions that are context-dependent. 123 Increased microglia senescence occurs in the aging brain,110,122 and microglia with disease-associated transcriptomic signatures also have a senescent transcriptomic phenotype. 124 Whole body elimination of senescent cells in aged mice using a genetic/pharmacologic INK-ATTAC strategy or a senolytic drug treatment regimen resulted in a selective decrease of p16INK4a + microglia, and improved cognitive function of aged mice, further supporting that senescent microglia can drive age-associated cognitive decline. How senescence of NVU cells influences BBB functions has not yet been formally addressed but could plausibly contribute to aspects of BBB dysfunction through at least two mechanisms: (1) the loss of protective interactions with BECs and (2) the toxic contribution of the SASP (Figure 2).
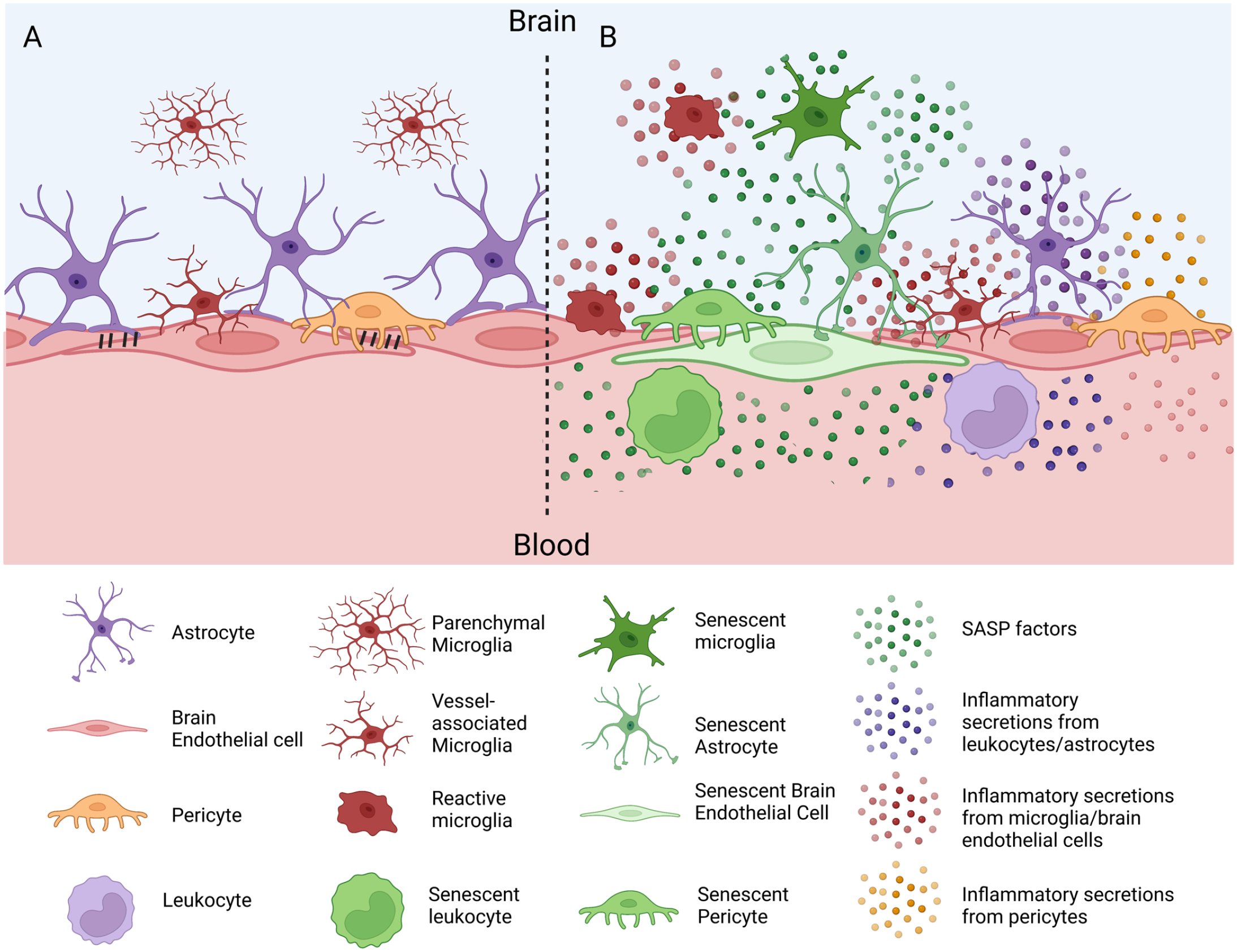
Cellular senescence and its possible contributions to BBB dysfunction. (A) Depiction of a healthy BBB with intact tight junctions and physical associations of BECs with astrocyte endfeet, pericytes, and microglia. (B) Changes of BBB function as a result of cellular senescence. Cellular senescence may occur in endothelial cells (green), resulting in cell-autonomous loss of BBB properties. Senescence of BBB-associated astrocytes, pericytes, and microglia (green cells) could contribute to loss of BBB maintenance/preservation following inflammatory insults. Senescent leukocytes, and particularly monocytes/macrophages (green), may also interact with the BBB or traffic across into brain parenchyma to induce neuroinflammation. The SASP from senescent cells in the brain, circulating leukocytes, or peripheral organs could lead to increases in SASP proteins (green dots) in brain ISF or blood that directly alters non-senescent brain endothelial cell functions, resulting in increased leakiness and leukocyte trafficking (purple cell). SASP proteins could also interact with BBB-associated cell types, altering their BBB-maintenance functions, or modulating their secretory phenotype. An increase in the secretion of inflammatory proteins by non-senescent pericytes (orange dots), astrocytes (purple dots, brain side), endothelial cells (pink dots), microglia (red dots), or monocytes/macrophages (purple dots, blood side) may contribute to the senescence-associated inflammatory milieu/localized reactive gliosis, and propagate BBB dysfunction. This figure was made with BioRender. BBB: blood–brain barrier; BEC: brain endothelial cell; SASP: senescence-associated secretory phenotype; ISF: interstitial fluid.
In addition to CNS cells of the NVU that may contribute to BBB dysfunction, senescence of cells that lie outside of the brain could also impact various functions of the BBB. For example, SASP factors can enter the blood stream as secreted proteins or packaged in exosomes. 125 Thus, an SASP from peripheral cells that are distant from the brain could interact with the luminal surface of BECs, affecting BEC function. A subset of p16INK4a positive infiltrating brain myeloid cells, together with senescent microglia, contributes to age-associated leukocyte trafficking across the BBB into the brain. 126 Systemic elimination of these senescent myeloid cells was associated with improved nest building in aged mice. 126 In summary, endothelial cells of the BBB and associated cells of the NVU can adopt senescent phenotypes with aging and age-associated diseases. However, to what extent cellular senescence within the BBB/NVU/CNS contributes to BBB dysfunction versus cellular senescence at distal sites in blood or peripheral organs remains unclear.
Cellular senescence as it relates to BBB dysfunction in peripheral/systemic diseases
Diabetes/obesity
Diabetes and obesity can elicit many stress pathways causing premature cellular senescence. As recently reviewed, there are multiple metabolic drivers of senescence that fit in with aging including mitochondrial dysfunction, redox imbalances, and hyperglycemia. 127 Exposure of endothelial cells to high glucose alone can trigger a senescent phenotype. 128 In addition, retinal endothelial cells undergo premature senescence as measured by SA-β-gal positive staining following exposure to high glucose. 129 Since senescence of cells can elicit a secretory phenotype, senescence of any cell within the NVU could contribute to the neurovascular inflammation associated with AD.
We have previously shown in a diabetic mouse model that treatment with a mitochondrial carbonic anhydrase inhibitor decreases oxidative stress and prevents BBB disruption and diabetes-induced pericyte loss at the NVU.130,131 The impact of this inhibitor on cellular senescence remains to be investigated. However, as carbonic anhydrases are increased with aging in brain 132 and with diabetes in heart, 133 targeting this enzyme would decrease oxidative stress and mitochondrial dysfunction, which could slow senescence.
While it is known that metabolic disease can induce many of the same stress pathways at the BBB that often lead to senescence of cells,134–136 whether senescence of BBB cells occurs in metabolic diseases has not been largely investigated. The blood–retina barrier (BRB) is a window into the BBB. Studying the BRB, it was found that arginase 1 is a contributor to diabetes-induced senescence of endothelial cells.
137
Arginase is an enzyme that uses
Peripheral senescent cells can promote insulin resistance, suggesting senescent cells can drive metabolic disease. 138 In either high-fat diet-induced obesity or in a genetic obese model (db/db mice), levels of senescent cells predominantly in the visceral adipose tissue were increased compared to controls. Senescent cell clearance through dasatinib and quercetin treatment improved glucose tolerance and insulin sensitivity but had no impact on body weight. As stated previously, due to the SASP, senescence in any peripheral organ could affect the BBB due to contact through the circulation.
Regular exercise can slow endothelial cell senescence in humans. 139 In endothelial cells collected from the forearm in young sedentary, aged sedentary, and aged individuals who exercised, protein levels of p21 and p16 were increased with age, with no age-related increases in the exercised individuals. Levels of these proteins were also inversely correlated with vascular endothelial function. These data suggest aerobic exercise may be able to protect against endothelial dysfunction that occurs with age by preventing endothelial cell senescence.
Inflammatory diseases
Chronic inflammation slows tissue remodeling and is often accompanied by cellular senescence. Inflammatory diseases that are organ-specific (e.g. originating in the lung like asthma, pneumonia, and COVID-19) as well as systemic diseases (e.g. arthritis) can contribute to vascular senescence. Because all organs are connected by the circulatory system, changes in one organ or cell type could impact other organs or cell types through endocrine signaling.
SARS-CoV-2 is an exemplary viral infection of the respiratory tract that has affected most of the world’s population. While SARS-CoV-2 primarily targets the lungs, a cytokine storm accompanying infection dramatically elevates many cytokines in blood, thus affecting every organ within the body.140,141 We have shown that a fragment of the viral attachment protein of the SARS-CoV-2 virus, S1, can cross the BBB. 142 There are also multiple different ways SARS-CoV-2 can have a detrimental impact on the BBB. 143 Increased oxidative stress and release of cytokines are two ways that the virus could directly impact the BBB. Whether infection increases senescence in BECs is still unknown. However, in a Nature Aging publication earlier this year, it was shown that SARS-CoV-2 infection can induce senescence of neighboring, uninfected cells in a paracrine manner. 144 Administration of dasatinib plus quercetin to rodents infected with SARS-CoV-2 decreased levels of inflammatory factors classified as the SASP.144,145 Therefore, it is plausible that SARS-CoV-2 could induce BBB dysfunction directly by inducing senescence of BECs or indirectly through senescence of adjacent cells or through systemic effects of the SASP.
During systemic inflammation, multiple studies have shown disruption of BBB integrity or enhanced BBB transport of inflammatory factors.146–148 Beyond the vagal afferents and choroid plexus, BECs are the first CNS cell type exposed to the damaging effects of peripheral inflammation. Inflammatory factors can trigger MMP secretion, endoplasmic reticulum stress, and mitochondrial damage. Gastrointestinal disorders not only release inflammatory cytokines but also alter the gut microbiota which can impact the BBB. 149 Thus, various peripheral inflammatory diseases can impact cellular senescence at the BBB.
Cancer
While this review focuses on the unfavorable aspects of cellular senescence, we must acknowledge its biological advantage as a tumor suppressant. Cellular acquisition of a fully senescent phenotype prevents premalignant and malignant tumorous cells from undergoing cell-cycle division. Moreover, the adoption of an SASP phenotype can act on neighboring cancer cells, improving the vasculature for drug delivery and recruiting immune cells to the cancerous region. 150 Many chemotherapeutics have been shown to stimulate therapy-induced senescence (TIS) such as doxorubicin, etoposide, and cisplatin.118,151 TIS cells have been identified in tumors following radiation or chemotherapy, 152 and are known to secrete the inflammatory cytokine interleukin 1α (IL-1α), a crucial SASP initiator and regulator. 152
However, it is also known that 17–75% of cancer victims treated with various chemotherapeutics, including doxorubicin, acquire chemotherapy-induced cognitive dysfunction (CICD) or “chemobrain.” 153 CICD is a syndrome of cognitive impairment that can persist long after treatment, and likely has BBB involvement. 154 TIS cells could be contributing to this debilitating phenotype. Adding to the complexity, it is known that TIS cells can induce ECM cleavage and growth factor release, which can promote tumorigenesis and eventual metastasis. 150 Thus, it is difficult to identify definitively whether senescent cells are pro-tumorigenic or antitumorigenic. Current work is investigating treatment paradigms of TIS coupled with senolytics which could halt tumor progression and provide an opportunity for senescent cell clearance. 150
Systemic cancer cells release IL-6, a key component of the SASP. This can not only disrupt the BBB but also crosses the intact BBB,155,156 thus participating in a neuro-immune axis. 20 If this disruption of the BBB is prevented by eliminating IL-6, the host can live longer with the tumor. 157 Tumor-bearing mouse hosts have increased systemic IL-6. When given antibodies directed at the IL-6 receptor, BBB disruption is prevented and lethality is decreased, despite no change in tumor burden. Similar results were obtained in a Drosophila tumor-bearing model. 157 These data suggest that morbidity in cancer can be improved by targeting downstream effects of inflammatory cytokines like IL-6, which is a predominant SASP factor.
Conclusions
The field of cellular senescence is rapidly expanding. Preclinical studies have demonstrated that senolytics have efficacy in the brain, but the specific cellular targets are unknown.158–160 In this review, we have presented evidence of normative age–associated changes to the BBB and carefully analyzed how some of these changes are reminiscent of cellular senescence. We have also considered age-associated disease in the brain and the periphery, and considered how these changes could induce cellular senescence at the BBB and in particular BECs. Finally, we have examined multiple in vitro and in vivo studies that induce cellular senescence in BBB model systems. Importantly, the hallmarks of aging are intertwined; for example, cellular senescence can be induced by telomere shortening and DNA damage. 2 Moreover, cellular senescence can cause stem cell exhaustion and altered intracellular communication. We have concentrated on cellular senescence, but recognize that some of the age-associated changes in BBB functions that we highlighted could be due, in part, to other hallmarks of aging.
While we have presented multiple studies evaluating BECs and BBB-associated cells like pericytes and astrocytes, more work is needed. Cellular senescence can take anywhere between three and seven days in vitro or ten days to six weeks in vivo to establish. However, it is unknown if the development and progression of the phenotype over time cause different changes to BBB function (e.g. the acute vs chronic phenotype). Furthermore, BECs can induce senescence in nearby cells and adjacent cells can induce senescence in BECs, but it is still not clear what cells or induction mechanism impacts normative age–related BBB changes. With the emergence of clinical trials investigating senolytics, more work is urgently needed to understand the importance and roles of cellular senescence at the BBB. It is still not known if cellular senescence is maladaptive or adaptive, and if it varies by tissue or induction mechanism. The review presented here seeks to highlight the complexities of cellular senescence on the BBB and encourage more studies on this intriguing topic.
Footnotes
Authors’ Contributions
All authors participated in the conceptualization, writing, and revision of this publication.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Biological Mechanisms of Healthy Aging Training Program NIH T32AG066574 (R.C.K.), the Joe W. & Dorthy Dorsett Brown Foundation (M.A.E), P30 DK017047-44 (E.M.R.), P30 AG066509 (E.M.R.), the Veterans Administration (W.A.B.), and R03AG051071 (M.J.R.) and R21AG073676 (M.J.R.).