
Abstract
Recent reports revealed that severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-infected patients can develop bacteremia; however, the circulating bacterial profile is not well studied. Therefore, this study has aimed to investigate circulating bacterial profile in mild (n = 15) and severe (n = 13) SARS-CoV-2-infected patients as well as healthy controls (n = 10), using 16S rDNA (V4) sequencing approach. The alpha diversity indexes and Bray–Curtis dissimilarity matrix revealed that the bacterial profiles between the two conditions are significantly different. Correspondingly, the relative abundance indicates that the predominant bacterial phylum in both conditions was Proteobacteria. At genus level, the dominant bacterial genera in the mild patients belonged to Sphingomonas, Stenotrophomonas, and Achromobacter, while bacterial genera belonging to Enhydrobacter, Comamonas, and Acinetobacter were dominant in the severe patients. Furthermore, Linear discriminant analysis (LDA) Effect Size (LEfSe). revealed that Stenotrophomonas, Delftia, Achromobacter, and Neisseria were enriched in the mild condition, while Agrobacterium, Comamonas, Pseudomonas, Corynebacterium, Alkaliphilus, and Kocuria were enriched in the severe patients. These results revealed a distinct circulating bacterial profile in the mild and severe SARS-CoV-2-infected patients, which may provide an insight for further therapeutic strategy.
Impact Statement
The present study focuses on the investigation of circulating bacterial profiles in both mild and severe SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2)-infected patients along with healthy control cohort to assess the severity during the infection by using the amplicon-based metagenomic approach, in which the sequencing approach can address the bacterial profile in blood circulation with both high sensitivity and high specificity, while also overcoming the limitation of bacterial culture method for unculturable organisms. Furthermore, the present findings provide an insight into whether severity during infection may be associated with differences in circulating bacterial profile as well as identifying potentially causative pathogens related to the development of bacteremia during SARS-CoV-2 infection.
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), also known as coronavirus disease-19 (COVID-19), was first reported in Wuhan, China. Subsequently, the World Health Organization (WHO) announced a pandemic on 11 March 2020. Bacterial co-infection is infrequent in SARS-CoV-2-infected patients during hospitalization (3–8%); however, this is more prevalent in intensive care unit (ICU) patients (14%).1 –3 Although bacterial co-infections are uncommon, the mortality rate can reach 57% in patients with bacterial co-infection. 1 In addition, the bacterial co-infection can develop to bacteremia, a contributing factor in 30–68% of the SARS-CoV-2-infected patients in ICU admission.1,4,5 Therefore, the investigation of circulating bacterial profiles may provide an insight into both identifying the potentially causative pathogens of bacteremia, which are usually overlooked, and also the differentiation of mild and severe cases of SARS-CoV-2-infected patients, based on circulating bacterial profiles.
Although bacterial culture is the gold standard in pathogen identification, this technique has many limitations, including the inability to identify uncultured bacteria and its time-consuming procedure. 6 The advancement in metagenomic technologies can provide a potential for identifying uncultured bacteria as well as offering time efficiency with promising outcomes (high specificity and sensitivity). 7 In this study, we aim to investigate the circulating bacterial profiles, determine the association of circulating bacterial profiles with severity in both mild and severe SARS-CoV-2-infected patients, using the 16S rDNA sequencing approach, and further address the potential causes of bacteremia, which are usually overlooked.
Materials and methods
Study cohort
In this retrospective observational study, Thai patients admitted to King Chulalongkorn Memorial Hospital in Bangkok, Thailand, were tested for SARS-CoV-2 infection using reverse transcription-polymerase chain reaction (RT-PCR) assay of a nasopharyngeal swab specimen. Then, the confirmed cases of SARS-CoV-2-infected patients were classified as having either a mild condition (n = 15), in which the patients were admitted to the In-Patient Department (IPD), or a severe condition (n = 13), where patients who showed evidence of pulmonary infiltration from chest radiography or chest computer tomography were considered as severe cases, and were sent to the ICU to receive mechanical ventilation. In addition, healthy adult individuals (n = 10) who had no infectious signs or history of hospital admission in the previous six months were recruited for blood collections and used as an experimental control. The blood of the patients in all groups was collected before treatment and immediately on the first day of admission and stored at −80°C for further processing. The clinical parameters were obtained by routine laboratory investigations (Table 1) and collected by the Department of Laboratory Medicine, King Chulalongkorn Memorial Hospital, Thai Red Cross Society, in accordance with ISO 15189 (4006/47).
Baseline characteristics.
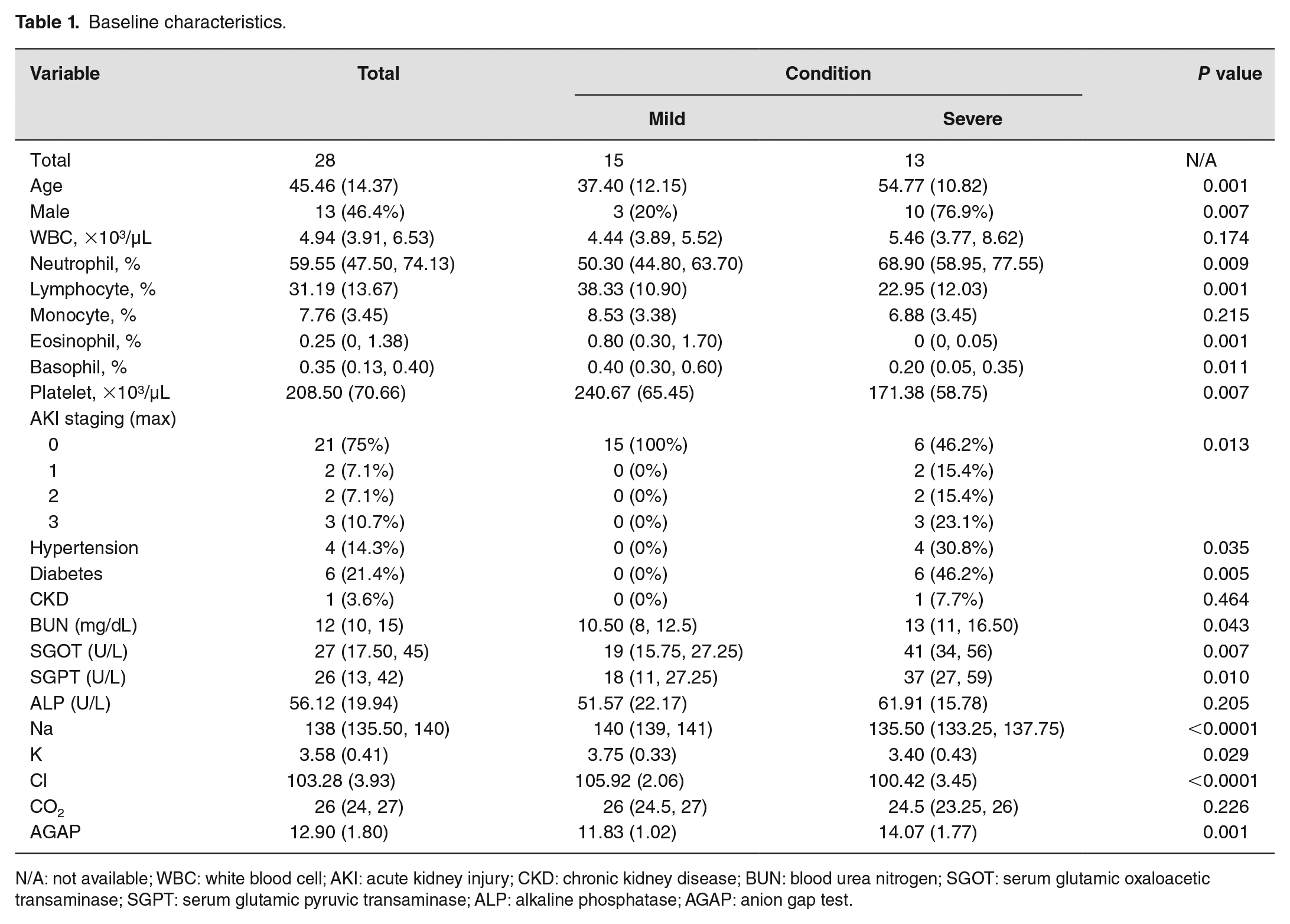
N/A: not available; WBC: white blood cell; AKI: acute kidney injury; CKD: chronic kidney disease; BUN: blood urea nitrogen; SGOT: serum glutamic oxaloacetic transaminase; SGPT: serum glutamic pyruvic transaminase; ALP: alkaline phosphatase; AGAP: anion gap test.
16S rDNA sequencing
For 16S rDNA sequencing, 300 μL of collected blood from patients was used for genomic DNA extraction with GenUp™ gDNA kit (Biotechrabbit, Germany), and PCR amplification was performed for the V4 region of 16S rDNA. The V4 amplicons were re-amplified with phasing adaptor primer and then sequenced with Illumina MiSeq platform (paired-end, 2×250 cycles). 8 The raw FASTQ files obtained from this study were deposited in the Sequence Read Archive (SRA) database under the BioProject (PRJNA783167) with the BioSample for mild (SAMN2356002) and severe (SAMN23425330) conditions.
Data analyses
The chi-square or Fisher exact test and the independent t test or Mann–Whitney U test were used to evaluate the clinical characteristics of the patients, and the results were reported as counts (percentages) for categorical data and median with interquartile range for continuous data. All statistical analyses for the clinical characteristics were performed using Stata version 15.1 (STATA Corp, TX). The P value less than 0.05 was considered statistically significant for all tests.
To investigate the circulating bacteria, the raw sequencing data sets were analyzed with QIIME2 pipeline (version 2020.8). 9 Then, the paired-end sequences were merged and trimmed based on quality score (<Q30), and the merged reads were deduplicated and clustered with 98% similarity using VSEARCH algorithm. 10 The UCHIME algorithm was applied to filter the chimeric sequences. 11 Subsequently, the filtered reads were annotated with the Greengenes database (version 13.8) using the VSEARCH algorithm.10,12 The alpha diversity indexes and principal coordinate analysis (PCoA) based on a Bray–Curtis dissimilarity matrix with permutational multivariate analysis of variance (PERMANOVA) were performed in R (version 4.1.0). Linear discriminant analysis (LDA) Effect Size (LEfSe) was introduced for differential abundance analysis. The significant difference was defined as P < 0.05, and the threshold on the logarithmic LDA score for the analysis was 4.0. 13
Results
According to the baseline characteristics (Table 1), 13 out of 28 patients were categorized as patients in severe condition, following the criteria described above. Among the characteristic features, the patients in a severe condition tended to be older, and with a higher male proportion, than those in the mild condition (P < 0.05), and some of the patients had comorbidity (e.g. diabetes, hypertension, and chronic kidney disease).
To investigate the circulating bacterial profile, the 16S rDNA sequencing was introduced. The results of the rarefaction curve analysis (Supplementary Figure 1) revealed that the sequencing depth was sufficient to capture the entire circulating bacterial taxa of the patients. The alpha diversity indexes (Figure 1(a)) of the severe condition were significantly higher than the mild condition (P < 0.05) and healthy controls (P < 0.01). In addition, the healthy control group revealed a similar pattern of Chao1 to that of patients with a mild condition (Figure 1(a)). These findings indicated that the severe SARS-CoV-2-infected patients had more circulating bacterial diversity than the mild SARS-CoV-2-infected patients in the manner of richness and evenness. Similarly, the analysis of the beta diversity index using Bray–Curtis dissimilarity (Figure 1(b)) revealed that the circulating bacteria of the severe SARS-CoV-2-infected patients were significantly different from both mild SARS-CoV-2-infected patients and healthy controls (PERMANOVA, P < 0.001).
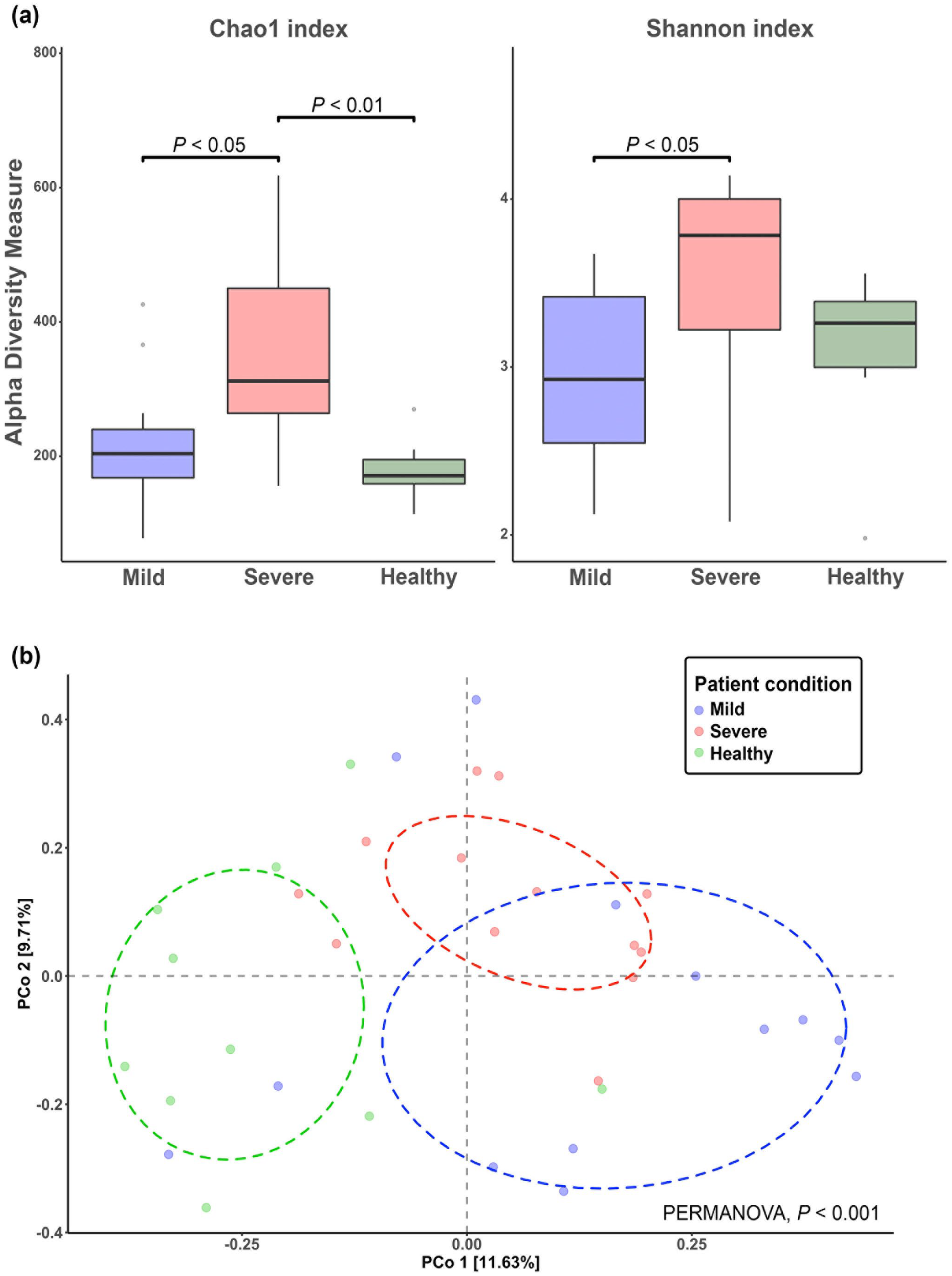
Alpha and beta diversity indexes of circulating bacteria in healthy control, mild, and severe conditions: (a) the Chao1 and Shannon indexes and (b) PCoA plot of beta diversity based on Bray-Curtis dissimilarity matrix.
The predominant bacterial phylum was Proteobacteria in both mild and severe SARS-CoV-2-infected patients, and also in healthy controls, as shown in Figure 2. However, the relative abundance of circulating bacteria at the genus level showed that the predominant taxa among groups were different (Figure 2(a)). For patients with a mild condition, the dominant bacterial genera were Sphingomonas followed by Stenotrophomonas and Achromobacter. Meanwhile, bacterial genera belonging to Enhydrobacter, Comamonas, and Acinetobacter were dominant in the SARS-CoV-2-infected patients of the severe condition, and the relative abundance of circulating bacteria of healthy controls revealed that the dominant bacterial genus was Bradyrhizobium (Figure 2(a)).
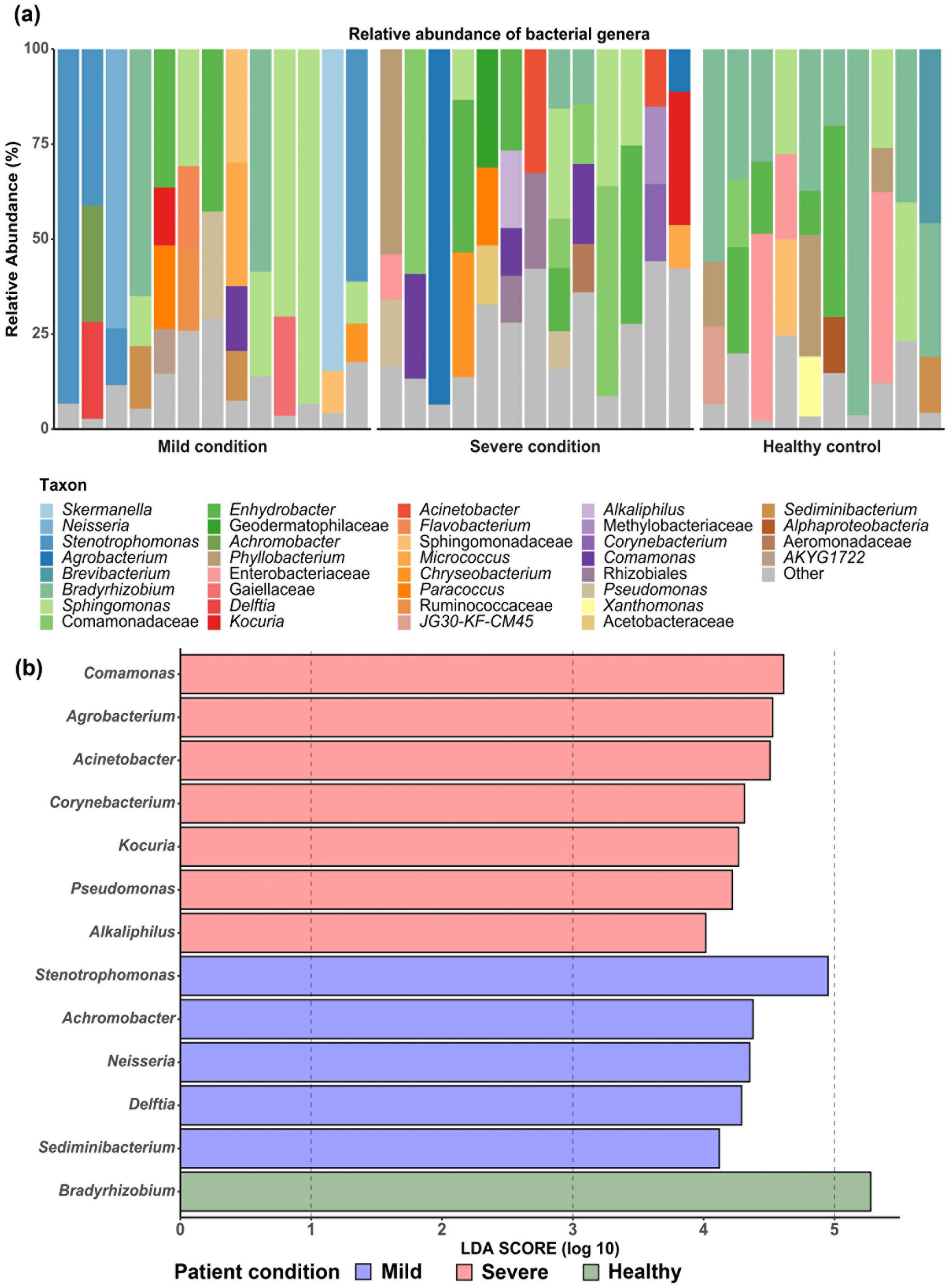
The circulating bacterial compositions in healthy control, mild, and severe conditions: (a) the relative abundance at the genus level and (b) Linear discriminant analysis (LDA) Effect Size (LEfSe) showing significant different bacteria among groups.
To examine whether the severity of infection in the SARS-CoV-2-infected patients was associated with differences in bacterial taxa, the LEfSe was conducted for differential abundance analysis among patients with mild condition, severe condition, and healthy control. 13 Then, the significant taxa from the LEfSe (Figure 2(b)) were selected based on previous reports as well as their relative abundance (>1%) in order to present only those bacterial genera potentially causative of the severity. The bacterial genera which were significantly enriched in each of the conditions is shown in Table 2. For the mild condition, the enriched circulating bacterial genera were Stenotrophomonas, Delftia, Achromobacter, and Neisseria. Meanwhile, the bacterial genera of Agrobacterium, Acinetobacter, Comamonas, Pseudomonas, Corynebacterium, Alkaliphilus, and Kocuria were enriched in the severe condition.
The potentially causative bacterial genera causing bacteremia from the result of the LEfSe.

LEfSe: Linear discriminant analysis Effect Size; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2.
The parenthesis behind the genus indicated the relative abundance belonged to the genus and N/A was abbreviated for not available data.
Discussion
Previously, gut and respiratory microbiota of SARS-CoV-2-infected patients were intensively investigated, revealing that the microbiota profile was associated with the infection. However, the association of COVID-19 severity, pathogenesis, and the human microbiome remains uncertain.14,15 Therefore, this study focused on an investigation of the relationship between circulating bacteria and severity in both severe and mild SARS-CoV-2-infected patients. In this study, most patients with a severe condition had comorbidity and were mostly male and older than those with the mild condition. Recent reports have revealed that age and gender were associated both with the risk and severity of the SARS-CoV-2-infected patients and also with the occurrence of bacteremia in the patients. 16 Furthermore, even though the number of white blood cell counts in the two groups of patients were not different, the neutrophils of the patients with the severe condition showed significantly higher numbers than those of the patients with the mild condition (P < 0.01), as shown in Table 1. This might be related to the variation of circulating bacterial compositions in mild and severe conditions.17,18 Moreover, the low levels of lymphocytes, eosinophils, basophils, and platelets were consistent with a previous report in which a high neutrophil-to-lymphocyte ratio was associated with severity of the patients’ condition. 16
Interestingly, the results of bacterial profiles in mild and severe conditions are inconsistent with previous reports for the SARS-CoV-2-infected patients with bacteremia.19 –21 Of note, the most common isolated causative microorganisms of bacteremia in the studies belong to the genera of Escherichia, Staphylococcus, and Streptococcus. However, these studies applied the blood culture technique to determine bacterial composition in the blood circulation, in which the limitations in culture-based technique might miss some bacterial genera, especially with unculturable bacteria.22,23 Thus, this study used the advantages of the metagenomic approach to investigate the bacterial composition in the blood circulation of the SARS-CoV-2-infected patients, and discovered a different result, including the potentially causative bacteria that might be associated with the severity of the SARS-CoV-2 infection.
To address whether circulating bacterial profiles were associated with the SARS-CoV-2 infection or comorbidity status (e.g. hypertension and diabetes) as well as sex, we performed a statistical analysis based on the bacterial composition in the blood circulation of the severe patients (Supplementary Figure 2). The results suggest that the circulating bacterial profiles in the severe group were not influenced by these factors. However, given the limitations of our study, notably small sample size and single-center study, we suggest that further investigation based on circulating bacterial profiles of SARS-CoV-2 infection with and without comorbidity status as well as influence of sex, using a multi-center study, should be pursued.
In the severe group, 9 of 13 patients had subsequent infections (Supplementary Table 1). All of them had positive bacterial cultures from endotracheal suctions or sputum. One patient had Pseudomonas pneumonia after admission. Four patients had Klebsiella pneumoniae pneumonia after admission. The remaining patients had Acinetobacter baumanmii pneumonia around seven days after ICU admission. These patients with pneumonia development after the ICU admission accounted for around 69.23% of the severe cases and were mostly caused by nosocomial bacterial infection. Therefore, the investigation of circulating bacterial profile of the SARS-CoV-2-infected patients in both mild and severe conditions may offer new insights into the disease pathogenesis and provide novel prevention and treatment strategies.
The LEfSe analysis revealed that most of the enriched bacterial genera were typically found in the respiratory tract, gastrointestinal tract, skin, and hospital environment. Interestingly, most of the genera have been previously reported as microorganisms causative of bacteremia, except Alkaliphilus. Thus, the bacterial genus of Alkaliphilus should be further investigated to understand an association between disease and severity. Furthermore, the procedure of mechanical ventilation may be related to the enriched bacteria in the bloodstream through secondary infections in the severe condition. 24 The enriched Gram-negative bacteria including Agrobacterium, Comamonas, and Pseudomonas may contribute to the severity via an endotoxin-mediated pathway in the severe patients. 25 In addition, the enriched Gram-positive bacteria (i.e. Corynebacterium, Alkaliphilus, and Cloacibacterium) in the severe condition might also be associated with the severity of the SARS-CoV-2 infection through the development of sepsis in ICU patients. 26
In conclusion, the findings of this study provide the knowledge of circulating bacterial composition as a resource for further application in the treatment of the SARS-CoV-2-infected patients with bacteremia. There are several limitations of this study (e.g. small sample size, single-center study, only Thai ethnicity, and focusing only on circulating bacterial profile). Therefore, future studies should expand the study cohort and ethnicity, and also investigate other possible circulating microorganisms (e.g. virus, protozoa, and fungi) in SARS-CoV-2-infected patients in other countries to establish the association of the causative agents with the severity of SARS-CoV-2 infection.
Supplemental Material
sj-pdf-1-ebm-10.1177_15353702231157931 – Supplemental material for Comparison of circulating bacterial profiles between mild and severe COVID-19 patients
Supplemental material, sj-pdf-1-ebm-10.1177_15353702231157931 for Comparison of circulating bacterial profiles between mild and severe COVID-19 patients by Pavaret Sivapornnukul, Suwalak Chitcharoen, Vorthon Sawaswong, Sasipha Tachaboon, Janejira Dinhuzen, Nattachai Srisawat and Sunchai Payungporn in Experimental Biology and Medicine
Footnotes
Acknowledgements
The authors would like to thank the staff of the Excellence Center for Critical Care Nephrology (EC-CCN) and Emerging Infectious Disease (EID) unit, Faculty of Medicine, Chulalongkorn University, as well as all members of the Center of Excellence in Systems Microbiology (CESM) for all assistance. Finally, the authors would like to acknowledge the Department of Microbiology, Faculty of Medicine, Chulalongkorn University, Thailand, and Development and Promotion of Science and Technology Talents Project (DPST) for all support.
Authors’ Contributions
PS, SC, NS, and SP were responsible for study concept and design. ST and JD collected the clinical data and processed statistical data. PS, SC, and VS were responsible for the acquisition, analysis, visualization, and interpretation of data. PS and SC drafted the manuscript. SP and NS supervised this study. All authors contributed significantly to the revision of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
The study was reviewed and approved by the Faculty of Medicine, Chulalongkorn University ethics committee (IRB no. 336/63). The informed consent was waived due to the observational nature of the study.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The funding was supported by Tropical Medicine Cluster, Chulalongkorn University, the Jongkolneenithi foundation, the Medical Association of Thailand, Chulalongkorn University, and Thailand Science Research and Innovation Fund Chulalongkorn University (CU_FRB65_hea (27)_034_30_15).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.