
Abstract
Sarcoidosis is a granulomatous disease of unknown etiology, with limited therapeutic options. Chronic sarcoidosis can result in pulmonary fibrosis and can be lethal. Enhanced expression of pro-inflammatory cytokines, such as interleukin-17A (IL-17A), has been observed in sarcoid granulomas in humans. However, the role of IL-17A in the pathogenesis of chronic sarcoidosis or sarcoidosis-related pulmonary fibrosis and its potential therapeutic effects remain unclear. This study investigated whether IL-17A is critical in granulomatosis and its role in chronic inflammation in a profibrotic manner. Wild-type and IL-17A-knockout C57BL/6 mice were repeatedly challenged with heat-killed Propionibacterium acnes (PA) to induce sarcoidosis-like granulomata and sarcoidosis-related pulmonary fibrosis. Wild-type mice with granulomatosis were treated with anti-IL-17A antibody. Administration of PA enhanced the expression of IL-17A, granulomatosis, and fibrosis in mouse lungs after boost stimulation. Neither granulomata nor fibrosis were observed in IL-17A-knockout mice, even in the presence of interferon-γ enhancement. Neutralizing IL-17A antibody reduced inflammatory cells in bronchoalveolar lavage fluid and ameliorated both granulomatosis and fibrosis in sarcoidosis mice. In conclusion, our data demonstrate that IL-17A plays a critical role in PA-induced sarcoidosis-like inflammation in both granulomatosis inflammation and disease progression to pulmonary fibrosis, thus providing novel insights into the treatment of chronic sarcoidosis or sarcoidosis-related pulmonary fibrosis.
Impact statement
Pulmonary fibrosis–associated sarcoidosis is lethal and lacks better understanding. Our research investigated how chronic inflammation progressed into pulmonary fibrosis in mice, which is similar to disease progression in patients. Interleukin-17A (IL-17A) is elevated in both lung granulomatosis and pulmonary fibrosis, and the role of IL-17A is independent of interferon-γ. The significant role of IL-17A in Propionibacterium acnes–induced sarcoidosis inflammation provides insights into the therapeutic targets of sarcoidosis.
Introduction
Sarcoidosis is a systemic non-caseating granulomatous inflammatory disease that predominantly affects the lungs in over 90% of patients. 1 Granulomas are formed in the presence of several pro-inflammatory cytokines under antigen triggers. Along with the granulomata, fibroblasts and collagen encase around the lesion, and further development of fibrosis can destroy the lung structure, resulting in a fatal situation. 2 Although most cases of sarcoidosis are in remission, up to one-third of cases acquire chronic and progressive courses. Furthermore, pulmonary fibrosis following sarcoidosis has not been well-documented, and studies are lacking. 3 Lethal respiratory failure and pulmonary hypertension always result from pulmonary fibrosis in sarcoidosis and can be life-threatening. As the pathogenesis of sarcoidosis remains largely unknown, non-specific anti-inflammatory approaches are currently the main treatment for sarcoidosis. Corticosteroids are the first-line therapy, whereas antimetabolites are used as second-line therapy if patients are refractory to or intolerant to corticosteroids. 4 Therefore, a better understanding of sarcoidosis and novel therapeutic strategies is needed.
Lymphocytes are key effector cells responsible for inflammatory cytokine secretion and granulomatosis. T helper-1 (Th-1) cell polarization has been considered a classical theory for granulomatosis pathogenesis. Interferon (IFN)-γ, tumor necrosis factor (TNF)-α, and interleukin (IL)-12 facilitate macrophage and other inflammatory cell migration and accumulation. In recent years, evidence has suggested a role of T helper 17 (Th17) cells in sarcoidosis. Researchers have found that IL-17A-expressing CD4+ T lymphocytes or IL-17A+IFN-γ+ memory T cells and Th17-specific transcription factor RAR-related orphan receptor-γt are increased in bronchoalveolar fluid and peripheral blood of patients with sarcoidosis; in few instances, they indicate an active form of the disease.5,6 IL-17A+ cells are also suggested to be persistently present in patients with relapse. 7 All these data provide evidence of the critical role of IL-17A in sarcoidosis, but further mechanistic research is needed. Moreover, no evidence for the role of IL-17A in chronic sarcoidosis or sarcoid pulmonary fibrosis has been reported. Hence, it is unclear whether IL-17A is a persistent pathogenic factor in the progression of chronic disease.
In sarcoidosis, fibrotic changes are prominent along bronchovascular bundles and septal lines, paralleling the distribution of granulomatous inflammation. 8 The histological features of sarcoid pulmonary fibrosis are unique and distinct from the usual interstitial pneumonia (UIP) pattern. Furthermore, the pathogenesis of pulmonary fibrosis following chronic sarcoidosis is unclear and requires further investigation. The immune response biased from an initially Th-1-polarized to a Th-2-polarized was one of the explanations. The Th2 immune response is responsible for wound healing, and profibrotic cytokines, such as CCL18, lead to collagen deposition and fibrosis. However, no solid evidence has been found to confirm that Th2 cells are responsible for sarcoid pulmonary fibrosis, while some studies have shown no significant differences in Th1 and Th2 cytokines in patients with sarcoidosis from different stages. To date, it is unclear whether fibrosis in sarcoidosis is due to an extension of persistent inflammation or an underlying immunophenotype. IL-17A was detected at a later phase of sarcoidosis together with IFN-γ and IL-13.9,10 However, the role of IL-17A in the fibrotic stage of sarcoidosis is poorly understood.
Propionibacterium acnes (PA) has been isolated from sarcoid lesions, and PA genomes have also been detected in the lymph nodes of patients with sarcoidosis.11,12 PA-induced granulomatosis in mice is the most commonly used animal model for sarcoidosis. To investigate the performance of IL-17A in all stages of sarcoidosis and the efficacy of targeting IL-17A as a therapeutic method for sarcoidosis, we induced both granulomatosis and fibrosis in wild-type mice and IL-17A-deficient mice using PA. This study aimed to elucidate the role of IL-17A in different stages of sarcoidosis, especially its role in pulmonary fibrosis, in order to clarify its potential therapeutic effects in all aspects of this disease.
Materials and methods
Mice
Specific pathogen-free C57BL/6 female mice aged 8–10 weeks were purchased from the Animal Center of Peking University (Beijing, China). IL-17A-knockout mice with a C57BL/6 background were provided by Dr Iwakura (University of Tokyo, Tokyo, Japan) 13 and were maintained up to 8−10 weeks of age under specific pathogen-free conditions in an environmentally controlled clean room at the Center for Experimental Medicine of Chaoyang Hospital, Capital Medical University. All experimental and control mice were weight-matched, and their weights ranged from 20 to 25 g. The mice were sacrificed at each observational time point according to acceptable euthanasia guidelines using an intraperitoneal pentobarbital overdose. This study was carried out in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and was approved by the Animal Care and Utilization Committee of Capital Medical University. All animal studies were performed in accordance with ARRIVE guidelines.
Reagents and mouse model establishment
PA was obtained from the American Type Culture Collection (ATCC #6919; Manassas, VA, USA) and cultivated under anaerobic conditions. The PA colonies were washed twice and resuspended in phosphate-buffered saline (PBS). The PA suspension was heat-killed by autoclaving at 121°C for 20 min and maintained at −80°C prior to use.
Mice received an intraperitoneal injection of the PA suspension (0.5 mg of PA suspension or an equal volume of PBS as a control). On day 14, mice were challenged with a 0.5 mg PA suspension via the intratracheal route. Mice in the sarcoid-fibrosis group were challenged with an extra 0.5 mg heat-killed PA suspension intratracheally on day 28, as previously described. 14 Wild-type mice challenged with PA received an intraperitoneal injection of 100 µg rat anti-mouse IL-17A antibody or Rat IgG1κ isotype control (BD Pharmingen, San Diego, CA, USA).
Bronchoalveolar lavage and lung homogenates
Bronchoalveolar lavage (BAL) cells were collected by three times injection of 0.8 mL of sterile PBS according to the regimen used in this study. The total number of BAL cells was counted using a hemocytometer. Cell pellets were suspended in PBS and cytospun onto glass slides; 200 cells were counted for each sample for cell classification after hematoxylin and eosin (H&E) staining. Whole lung tissues were homogenized in cold lysis buffer (PBS containing 0.1% Triton X-100 with 0.1% protease inhibitor) and then centrifuged at 1500 ×g for 15 min at 4°C to obtain supernatants for cytokine analysis. 14
Histological assessment
The left lobes of the mouse lungs were inflated and then immersed in 10% buffered formalin for more than 24 h before paraffin embedding. The lungs were dehydrated, paraffin-embedded, and cut into 4-μm sections. Lung sections were stained using H&E or Masson staining. Immunostaining was performed using an antibody against IL-17A (Abcam, Cambridge, MA, USA; 1:100). The severity of pulmonary fibrosis was semi-quantitatively assessed using Ashcroft’s scoring system. The sections were assessed using an Olympus IX-51 microscope (Olympus, Tokyo, Japan), and images were obtained using an Olympus DP 70 camera (Olympus, Tokyo, Japan). Assessment of granuloma formation and immunostaining of cells were performed using the Image-Pro Plus software package version 6.0, and the granuloma areas were calculated by tracing individual borders in the software.
Cytokine expression analysis
Enzyme-linked immunosorbent assay (ELISA) was used to determine the IFN-γ, IL-17A, and transforming growth factor β (TGF-β) concentrations in mouse BAL and homogenates with respective ELISA kits purchased from eBioscience (San Diego, CA, USA) according to the manufacturer’s instructions.
Flow cytometry assay
Mouse lung cells were isolated as previously described. 15 They were stimulated for 4 h at 37°C with 0.25 μM Phorbol-12-myristate-13-acetate (PMA) and 1 μg/mL ionomycin (Sigma-Aldrich, St. Louis, MO, USA) in the presence of 10 µg/mL Brefeldin A (Enzo Life Sciences, Shanghai, China). The cells were stained with antibodies against CD3, CD8, CD4, and IFN-γ (BD Pharmingen). Flow cytometry was performed using a FACSCanto II (BD Biosciences, San Jose, CA, USA) and analyzed using BD FACSDiva Software (De Novo Software, Los Angeles, CA, USA).
Statistical analysis
Groups of four to six mice were used at each time point in each experiment. All data are expressed as mean ± SEM. All in vitro experiments were conducted in triplicate. GraphPad Prism 6 (GraphPad, La Jolla, CA, USA) was used for statistical analyses using Student’s t test or one-way analysis of variance for continuous variables. Statistical significance was set at P < 0.05.
Patient and public involvement
None.
Results
PA induce a sarcoid-like mouse model and chronic inflammation-associated pulmonary fibrosis
Sarcoid-like granulomas were induced in wild-type mice using intraperitoneal and then intratracheal PA injections. Figure 1(A) shows the regimen of the experiment. Mice boosted with PA on days 0 and 14 could induce a significant increase in IFN-γ and IL-17A levels in the lungs on day 21, and this inflammation response would resolve on day 28, with a decrease in cytokine levels. IL-17A and IFN-γ in BAL and lung homogenates were elevated along with the formation of granulomata, indicating the potential involvement of both IFN-γ and IL-17-associated responses in inflammation and granulomatosis. Two PA injection-induced granulomatosis inflammation (called sarcoid-granulomata) groups in mice matched some patients with sarcoidosis whose granulomata resolved without treatment. However, another boost of PA on day 28 helped to prolong the inflammation and IFN-γ and IL-17A influx for a second time, which lasted for a long time. At day 35, three PA boosts induced TGF-β elevation in the sarcoid-fibrosis group (Figure 1(B)). As three injections of PA induced continuous IFN-γ and IL-17A-related inflammation in mouse lungs, a complete process of sarcoidosis from granulomata to pulmonary fibrosis occurred as well. On day 21, the granulomata were significant, and chronic inflammation-induced pulmonary fibrosis was observed on day 63 (Figure 1(C)).
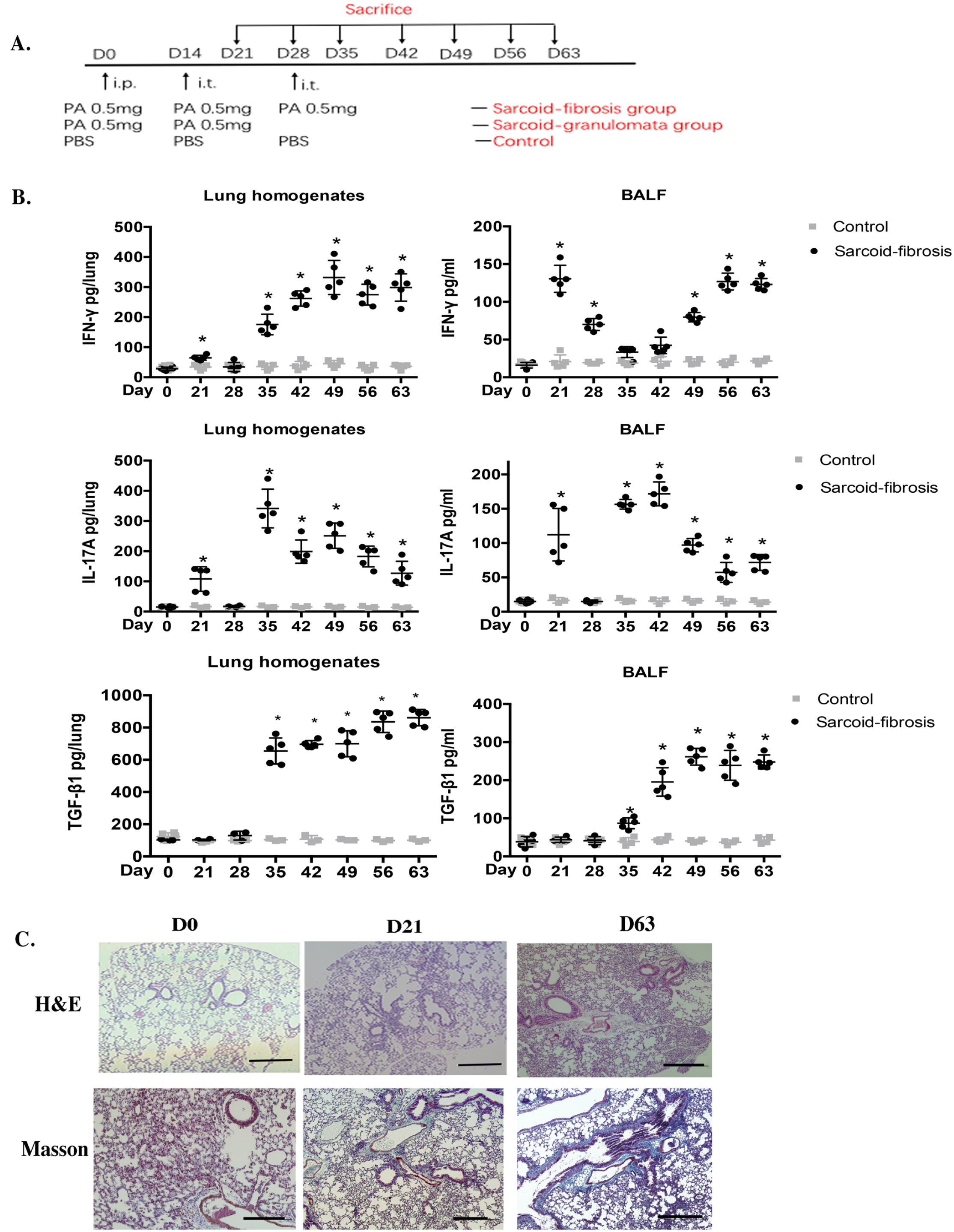
IL-17A is a key effector in PA-induced granulomatosis inflammation. (A) The experimental regimen of the sarcoidosis mouse model. i.p.: intraperitoneal; i.t.: intratracheal. (B) Protein levels of IL-17A, IFN-γ, and TGF-β in lung homogenates and BALF from day 21 to day 63. (C) H&E staining and Masson staining showed pulmonary fibrosis following chronic inflammation on day 63 in mouse lungs. ×10 magnification (bar = 200 µm). Data represent mean values ± SEM. *P < 0.05 compared with wild-type mice. Each group had four to six mice, and every in vitro experiment was repeated three times.
IL-17A is required for granulomatous inflammation in mice
H&E staining showed that lymphocytes and macrophages infiltrated mainly along the peribronchovascular regions and the interstitium of the lungs. Sarcoidosis-like granulomata were confirmed by loose granulomata that formed mainly along with peribronchovascular bundles on day 21 in wild-type mice and self-resolved on day 28 in the sarcoid-granulomata group.
To elucidate the therapeutic role of IL-17A in sarcoidosis granulomata, PA-induced granulomatosis was performed in IL-17A knockout mice. H&E staining showed no obvious histological granulomata in IL-17A knockout mice on day 21. To determine whether inflammation was suppressed or delayed by IL-17A inhibition, we performed extended histological observation of the mice in the sarcoid-granulomata group. From day 28, inflammatory cell infiltration was resolved in both wild-type mice and IL-17A-knockout mice. On day 35, sarcoid-granulomatous inflammation was almost abolished but not delayed in the sarcoid-granulomata group by two PA injections (Figure 2(A)).
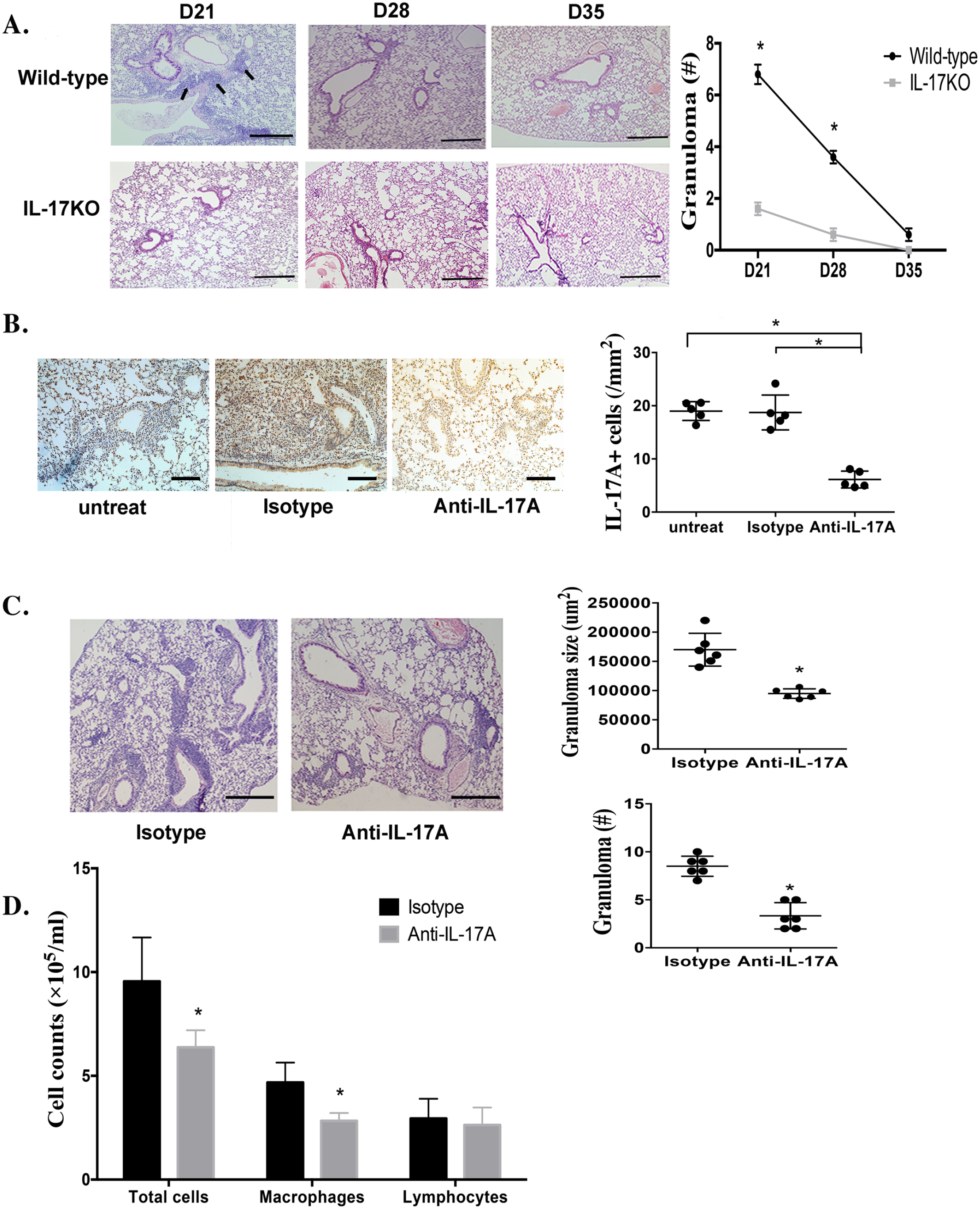
Anti-IL-17A antibody relieves granulomatous inflammation in mice challenged with PA. (A) H&E staining on the lungs of C57BL/6 wild-type and IL-17A knockout mice after PA challenge twice resolved on day 21, 28 and day 35. Arrows indicate granulomata along the peribronchovascular regions in wild-type mice. ×10 magnification (bar = 200 µm). (B) Immunohistochemical staining of IL-17A in lung tissue of PA-challenged mice on day 21 after anti-IL-17A antibody treatment. Right: average counts of IL-17A+ cells per unit. Sections are shown at a magnification of ×40 (bar = 50 µm). Data represent mean values ± SEM. *P < 0.05. (C) The lung histopathology of PA-challenged mice with anti-IL-17A antibody treatment was evaluated on day 21. H&E-stained sections are shown at magnifications of ×10 (bar = 200 µm). Granuloma number and size determined in the respective sections were compared between the anti-IL-17A and IgG isotype groups. Data represent means values ± SEM. *P < 0.05. (D) Inflammatory cells were present in the BALF from mice with anti-IL-17A treatment on day 21. Data represent mean values ± SEM. *P < 0.05. Each group had six mice, and every in vitro experiment was repeated three times.
IL-17A was detected as a pro-inflammatory factor in granulomatous inflammation based on the above data, which was accompanied by inflammatory cell infiltration and granuloma formation histologically. To determine whether blockade of IL-17A could inhibit pulmonary granulomatous inflammation, anti-IL-17A antibody treatment was administered every other day from day 15 to day 19, immediately after intratracheal PA injection. IgG isotype antibodies were used as controls. A group of mice treated with an equal volume of sterile PBS was set as the untreated blank control. All mice were sacrificed on day 21. Immunostaining of IL-17A expression in the lung showed fewer IL-17A+ cells after anti-IL-17A antibody treatment than in the control groups (Figure 2(B)). Histological analysis revealed significant reductions in both the size and number of granulomas in the lungs after IL-17A inhibition (Figure 2(C)). Moreover, total BAL cell counts and macrophage numbers were significantly reduced after IL-17A inhibition (Figure 2(D)). Taken together, IL-17A participates in granulomatous inflammation, and suppression of IL-17A may be a potential treatment for sarcoid-granulomata.
IL-17A is critical for pulmonary fibrosis following chronic granulomatous inflammation
Our previous study demonstrated that repeatedly challenging mice with PA would diminish granulomata in conjunction with lung fibrosis progression during periodic long-term observation, and significant sarcoid-fibrosis with little granulomata could be observed on day 63. 14 Based on this sarcoidosis-fibrosis mouse model, studies have been conducted to elucidate the role of IL-17A in chronic sarcoidosis. Anti-IL-17A antibody was thought to play a preventive role before PA boosting, and when pulmonary fibrosis became apparent on day 42, antibody treatment was considered the treatment group. The detailed regimen for the entire chronic sarcoidosis animal study is shown in Figure 3(A). Masson staining showed no lung fibrosis in IL-17A-knockout mice and significant alleviation of lung fibrosis in wild-type mice in the sarcoid-fibrosis group after anti-IL-17A treatment (Figure 3(B)). Consistent with the pathological findings, reduced hydroxyproline and mRNA levels of collagen I and III in the lungs also verified the diminishment of fibrosis after IL-17A inhibition (Figure 3(C)). Cells in BAL fluid were decreased after IL-17A inhibition under PA boost, especially macrophages, which were significantly decreased compared to the untreated sarcoid-fibrosis group (Figure 3(D)).
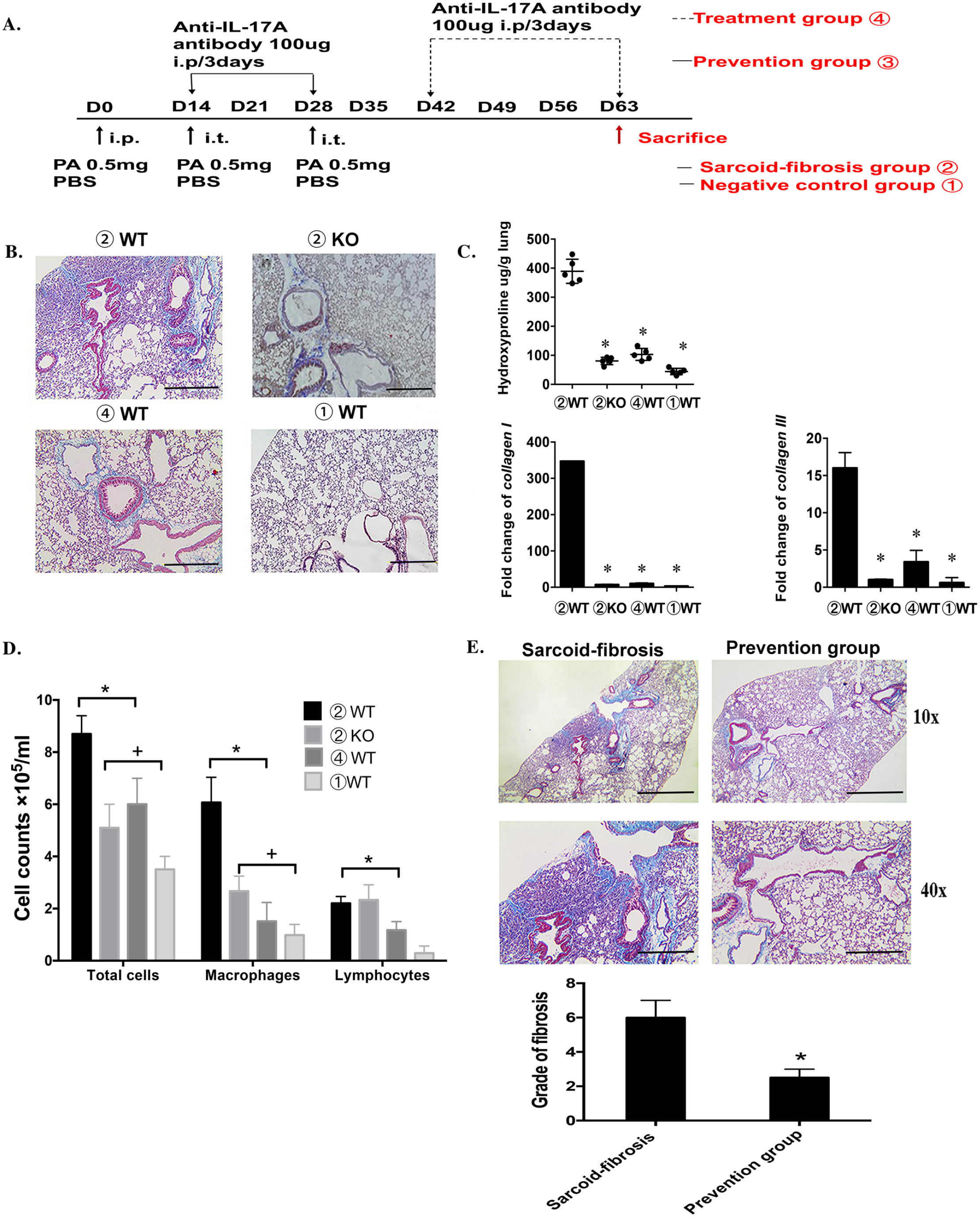
IL-17A is a therapeutic target for sarcoid-like pulmonary fibrosis. (A) The experimental schedules of sarcoid-fibrosis induction and IL-17A neutralization with IL-17A antibodies in mice. (B) The pathological evaluation of wild-type and IL-17A knockout mice with PA boosting; the treatment group stands for wild-type mice under PA boosting and provided an anti-IL-17A antibody. All the pathological evaluations are from the mice on day 63. Masson-stained sections are shown at magnifications of ×10 (bar = 200 µm). (C) The severity of fibrosis is evaluated by the level of hydroxyproline and the mRNA levels of collagen I and III in the lungs. Group numbers are labeled as shown in Figure 3(A). Data represent mean values ± SEM. *P < 0.05, compared with the negative control group.+P < 0.05, compared with the sarcoid-fibrosis group in wild-type mice. (D) Inflammatory cell distribution in the BALF in different sarcoid-fibrosis groups. The BAL was collected on day 63. (E) Masson-stained sections were shown at magnifications of ×10 (bar = 200 µm) and ×40 (bar = 50 µm) for mice in prevention groups. Ashcroft’s scoring for assessing fibrosis grade is shown in the right. Each group had six mice, and every in vitro experiment was repeated three times.
Moreover, IL-17A could also prevent disease progression; with anti-IL-17A antibodies and a third-time PA boost during the same period, pulmonary fibrosis was successfully avoided. Masson staining and Ashcroft’s scoring showed that the prevention group had a lower degree of fibrosis than the sarcoid-fibrosis group (Figure 3(E)), indicating the preventive role of anti-IL-17A antibodies in sarcoidosis-related pulmonary fibrosis.
IL-17 induced sarcoid-associated inflammation independent of IFN-γ and TGF-β
To investigate the potential interplay between Th-1 and Th-17 immune responses in PA-induced granulomatous inflammation, Th-1 cells and IFN-γ expression were evaluated. The ratio of IFN-γ+ CD4+ cells to CD4+ T cells was detected in the mouse lungs by flow cytometry analysis. No significant differences were observed in the proportion of CD4+ T cells between the wild-type and IL-17A-knockout mice (36.57 ± 2.67% vs 33.53 ± 2.08%). However, the frequency of IFN-γ+ cells was significantly increased in CD4+ T lymphocytes in the lungs of IL-17A knockout mice (8.03 ± 0.94 vs 31.5 ± 0.3%) (Figure 4(A)). Both IFN-γ mRNA levels and IFN-γ protein levels in lung homogenates and BAL were elevated in IL-17A-knockout mice compared with those in wild-type mice (Figure 4(B)). These data suggest an upregulation of the Th1 immune response in IL-17A knockout mice after PA administration. In the absence of IL-17A, the Th1 immune response alone could not induce granulomatosis inflammation.
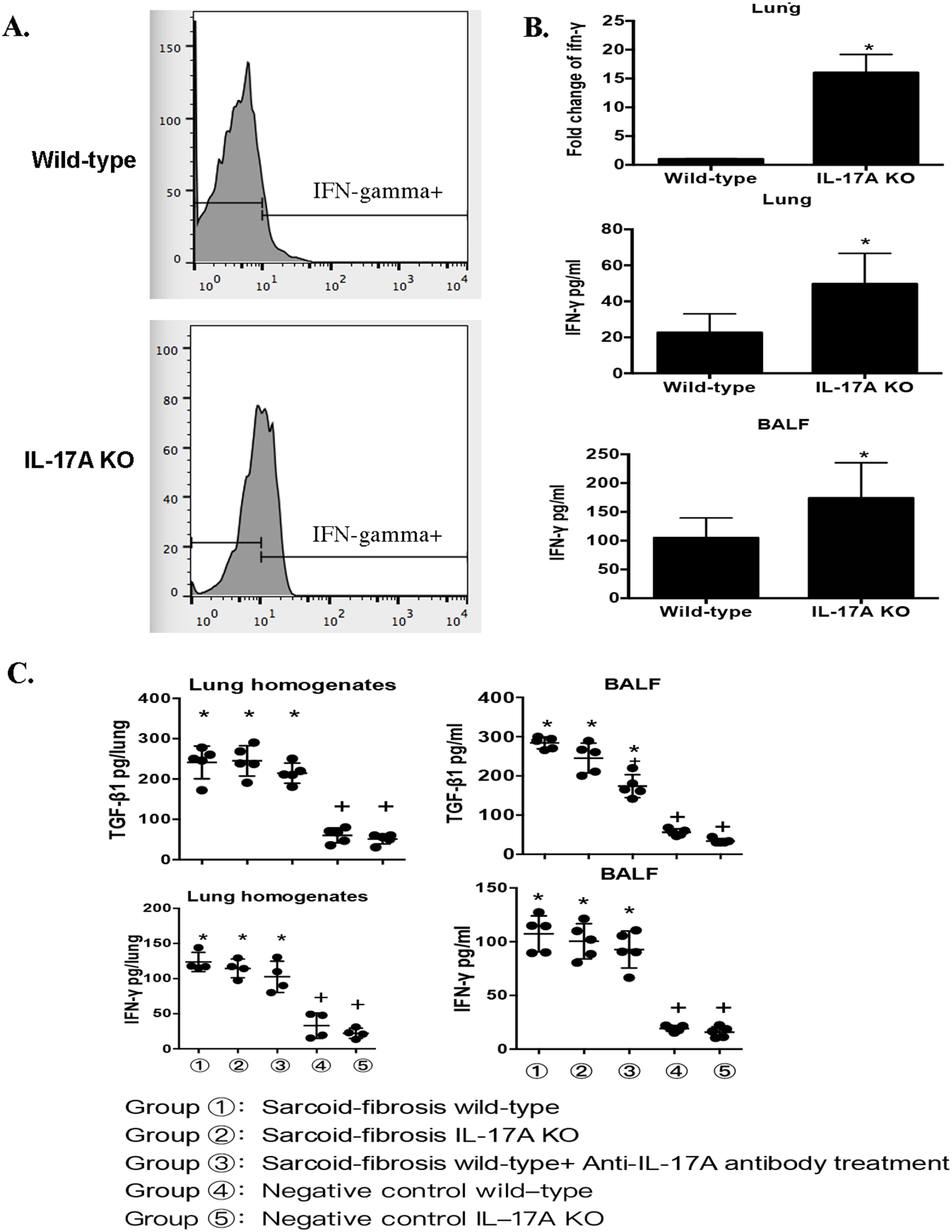
The immune response to sarcoidosis is dependent on IL-17A. (A) Flow cytometric characterization of IFN-γ+ CD4+ T cells in the lungs of PA-challenged IL-17 knockout and wild-type mice on day 21. (B) mRNA levels of IFN-γ in lung homogenates and protein levels in lung homogenates and lavage fluid on day 21 after PA challenge. (C) Protein levels of TGF-β and IFN-γ in lung homogenates and BALF in PA-induced sarcoid-fibrosis mice. Data represent mean values ± SEM. *P < 0.05, compared with the negative control of wild-type mice.+P < 0.05, compared with the sarcoidosis-fibrosis wild-type mice. Each group had four to six mice, and every in vitro experiment was repeated three times.
TGF-β and IFN-γ levels were both elevated in mice with sarcoid-fibrosis, as mentioned above. To investigate the relationship between IL-17 and profibrotic cytokines, the protein levels of TGF-β and IFN-γ were measured in mice in the sarcoid-fibrotic group. The expression of these cytokines was significantly increased in all mice under PA boosting compared to the negative control group. However, there were no differences between the sarcoid-fibrosis group and those with IL-17A deficiency. The data suggest that the increased expression of TGF-β and IFN-γ in lung fibrosis was independent of IL-17A levels (Figure 4(C)). Therefore, the data indicated that in the absence of IL-17A, TGF-β and IFN-γ upregulation could not induce lung fibrosis.
Discussion
IL-17+ cells and a higher expression of IL-17A have been previously reported in patients with sarcoidosis. Whether IL-17A plays a role in chronic sarcoidosis, especially pulmonary fibrosis, remains unknown, and the therapeutic role of IL-17A requires further investigation. In the present study, mouse models of PA-induced pulmonary sarcoid-granulomatosis and sarcoid-associated fibrosis were established to investigate the role of IL-17A in sarcoidosis at various stages. Pulmonary inflammation and fibrosis were both alleviated in IL-17A-deficient mice, and the anti-IL-17A antibody reduced pulmonary granulomatous inflammation and prevented disease progression in wild-type mice. We also demonstrated that in the absence of IL-17A, elevation of Th1 cells and IFN-γ could not induce granulomatosis inflammation or pulmonary fibrosis.
Th1 and Th17 cells are pro-inflammatory subtypes of CD4+ T cells and are involved in inflammatory and autoimmune diseases. A previous study demonstrated that PA-induced Th1-type responses led to granuloma formation in mice, 16 similar to the pathogenesis of human sarcoidosis. Some studies have attempted to find evidence of higher Th17 expression in sarcoidosis patients. They found that Th17 cells are specific for the early secretory antigenic target of 6kD (ESAT-6), which is an antigen from mycobacteria that may induce sarcoidosis. 17 Furthermore, a significant decrease in cytotoxic T-lymphocyte antigen 4 (CTLA4) expression in Th17 cells from mediastinal lymph nodes and BALF in sarcoidosis may contribute to Th17 priming and activation. 18 However, the relationship between Th17 cells and Th1 cells has not yet been elucidated. However, the therapeutic role of IL-17A in sarcoidosis requires further investigation. We found elevated IL-17A levels in sarcoidosis mice during the initial and granulomatosis inflammation phases, and a Th17-associated immune response in the pathogenesis of granuloma. In support of IL-17A involvement in sarcoidosis, enhanced IL-17A+ cells and IL-17A+IFN-γ+ memory T cells were detected in the blood and BAL of patients.12,19,20 However, in this study, in the absence of IL-17A, the proportion of IFN-γ+CD4+ T cells increased, with no granuloma formation. This result showed a significant role for IL-17A in granuloma formation.
Unlike idiopathic pulmonary fibrosis (IPF), sarcoidosis-associated pulmonary fibrosis occurs along with granulomata and is considered an extension of granulomatous inflammation. The pathogenesis of IPF and sarcoid pulmonary fibrosis is quite different; several well-defined factors, such as mucin 5 type B in IPF, are not associated with sarcoidosis. 21 Pathological and genetic studies of sarcoidosis suggest a pivotal role for inflammation and immune responses in sarcoid pulmonary fibrosis. 22 A better understanding of the pathophysiology of pulmonary sarcoidosis and the progression of granulomas to fibrosis is urgently needed. Most studies on sarcoidosis pathogenesis have so far focused on granulomatosis and inflammation. Song et al. 23 demonstrated Th17 elevation in Proprionibacteirum acnes-induced granulomatosis in a mouse model between days 15 and 56. In the present study, for the first time, exploring the progression of sarcoidosis from granulomatosis inflammation to pulmonary fibrosis, we described the role of IL-17A in sarcoid-granulomatosis-associated pulmonary fibrosis, which mimics chronic sarcoidosis in humans. IL-17 levels were elevated in these two distinct disease stages and exhibited both pro-inflammatory and profibrotic effects. IL-17A has been implicated in bleomycin-induced fibrosis in mice 24 and airway obstruction in chronic obstructive pulmonary disease in humans. 25 IL-17 signaling facilitates the production of IL-1β and TNF-α and increases the expression of TGF-β, which induces collagen I in mice. 26 However, the role of IL-17A in the pathogenesis of pulmonary fibrosis following sarcoidosis has not yet been studied in animal models or in patients. In IL-17A knockout mice, we found that a lack of IL-17A prevented chronic inflammation and progression to fibrosis, even in the presence of higher IFN-γ and TGF-β expression. To the best of our knowledge, this is the first report to explore IL-17 as a potential profibrotic cytokine, not only a pro-inflammatory cytokine, in pulmonary fibrosis following sarcoidosis.
Furthermore, we also demonstrated that the effect of IL-17A is independent of IFN-γ. Th-1-based inflammation is a well-known cause of sarcoidosis. In the present study, we suggested a novel theory that if IL-17 was lacking, exogenous antigen resulted in an increase in IFN-γ, and Th-1-based inflammation could not induce granulomatosis inflammation. After boosting the PA challenge, an enhanced Th1-polarized immune response was observed in the lungs, with a remarkable elevation in IFN-γ. In such inflammatory circumstances, treatment with anti-IL-17A antibody before PA boosting prevents mouse lungs from inflammation expansion and progression into a chronic course. In the present study, PA-induced Th1 and Th17 immune responses resulted in granulomatosis inflammation in the lungs. Enhanced IFNγ production was detected in the absence of IL-17A in PA-induced granulomata. Monocyte-induced IFN-γ is increased in sarcoidosis patients, while Th17 cells have been reported to decrease IFN-γ expression in another study. 27 A previous study in a mouse model of malignant pleural effusion revealed that IFN-γ expression was higher in IL-17a−/− mice. 28 The mechanism by which these two major cytokines function in sarcoidosis is unclear. Here, both mRNA and protein levels of IFN-γ were higher in IL-17A-knockout mice. In addition, the frequency of IFN-γ+CD4+ T cells was also increased in IL-17A-deficient mice. Our data suggested a negative interaction between IL-17A and IFN-γ. Granulomatous inflammation was significantly alleviated, and no typical granulomata were observed even with IFN-γ elevation in the lungs of IL-17A-deficient mice. These findings demonstrate the central role of IL-17A in granulomatous inflammation.
Studies in animal models of allergic airway inflammation have determined that treatment with an anti-IL-17A antibody could reduce the number of bronchoalveolar macrophages. 22 In our study, total cell counts, especially macrophage numbers in BALF, were significantly reduced in wild-type mice after IL-17A antibody treatment. IL-17A blockade effectively reduced inflammatory cell aggregates in the lungs and prevented immune responses. These findings were validated in IL-17A-knockout mice. Pathological analysis of BALF cells and lungs revealed an influx of macrophages after the PA challenge. The appearance of macrophages in both granulomatous inflammation and fibrosis suggests that macrophage-related chronic inflammation may be involved in sarcoidosis-associated fibrosis. The inhibition of IL-17A alleviates inflammatory cells, including macrophage accumulation, and prevents persistent inflammation. Both innate cells and adaptive T cells could be triggered by IL-17A amplification; 29 it has not yet been determined which cells play a major role in the sarcoidosis-associated IL-17A immune response.
In conclusion, recurrent PA stimulation can induce recurrence of sarcoidosis and subsequent fibrosis, in which IL-17A plays a critical role. We suggest that targeting IL-17A may represent a novel therapeutic strategy for sarcoidosis.
Footnotes
Authors’ Contributions
DJ and HD contributed to conceptualization; DJ, HX, and XZ contributed to investigation; RD contributed to resources; DJ contributed to writing—original draft preparation; HD and HZ contributed to writing—review and editing; HD contributed to project administration; DJ and HD contributed to funding acquisition. All the authors have read and agreed to the published version of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the National Natural Science Foundation of China (82170080); National High Level Hospital Clinical Research Funding, Elite Medical Professionals Project of China-Japan Friendship Hospital (ZRJY2021-GG11).