
Abstract
Neutrophil extracellular traps (NETs) are network-like structures of chromatin filaments decorated by histones, granules, and cytoplasmic-derived proteins expelled by activated neutrophils under multiple pathogenic conditions. NETs not only capture pathogens in innate immunity but also respond to sterile inflammatory stimuli in atherosclerosis, such as lipoproteins and inflammatory cytokines. Atherosclerosis is a lipid-driven chronic inflammatory disease characterized by the accumulation and transformation of inflammatory cells, and smooth muscle cells in the intimal space. NETs-derived extracellular components possess toxic and proinflammatory properties leading to cellular dysfunction and tissue damage, which may establish a link among lipid metabolism, inflammatory immunity, and atherosclerosis. In this review, we discuss recent advances regarding the role of NETs engaged in the pathogenesis of atherosclerosis, particularly focusing on the interaction with lipids and inflammasomes, crosstalk with smooth muscle cells and inflammatory cells, and the association with aging. We also evaluate the current knowledge on the potential of NETs as biomarkers and therapeutic targets for atherosclerosis and its related diseases in clinical practice.
Impact statement
In addition to capturing pathogens during innate immunity, neutrophil extracellular traps (NETs) respond to sterile inflammatory stimuli during atherosclerosis, such as lipoproteins and inflammatory cytokines. This review discussed recent advances regarding the emerging role of NETs in the pathogenesis of atherosclerosis, particularly focusing on their interaction with lipids and inflammasomes, crosstalk with smooth muscle cells and inflammatory cells, and the association with aging. We also evaluate the current knowledge about the potential of NETs as biomarkers and therapeutic targets for atherosclerosis and its related diseases in clinical practice.
Introduction
Despite declining death rates for cardiovascular disease (CVD) through current therapies, CVD remains the most common cause of death worldwide. 1 Atherosclerosis (AS) is the primary pathophysiology of CVD, which originates from a lipid-driven chronic inflammatory disease of the large arteries. 2 A long-term accumulation of plasma lipoproteins, namely hyperlipidemia, triggers endothelial cells (ECs) dysfunction, monocyte adhesion, entry into the intima, and differentiation into macrophages. In turn, macrophages take up the lipoproteins, including oxidized low-density lipoprotein (oxLDL) and triglyceride-rich lipoprotein remnants, giving rise to foam cells. Subsequently, migration, proliferation, and synthesis of extracellular matrix macromolecule in the resident smooth muscle cells (SMCs) are involved in lesion progression. The expanding foam cells and dead cells contribute to the formation of cholesterol-rich necrotic cores, which eventually may lead to clinical complications. The growing lesions will reduce blood flow leading to vessel occlusion, thus causing angina pectoris, especially during exercise or stress.3,4 Advancing lesions become susceptible to rupture following the expansion of necrotic cores and thinner fibrous caps, due to the participation of a series of inflammatory components. If the rupture occurs in the coronary artery, it can result in local thrombosis and even complete blood flow obstruction, generating myocardial infarction (MI). 5 Alternatively, the embolus can escape the heart and travel to the brain, where it may lead to stroke, or lodge in distal arteries, where it may lead to local ischemia, organ dysfunction, or potential infarction. 6 Although lipids undoubtedly contribute to AS, the accompanying inflammatory response dominates the progression and outcome. 7
Neutrophils are the most abundant inflammatory cells in the peripheral blood circulation, which play an antimicrobial role through phagocytosis and degranulation as first responders to acute inflammation. 8 Given their limited life span, neutrophils have long been underestimated in chronic inflammatory diseases, such as atherosclerosis, diabetes mellitus, and autoimmune disorders. Recently, a revival of neutrophils in chronic cardiovascular inflammation has aroused concern, 9 which results from the discovery of a third antimicrobial mechanism – NETs as an essential contributor to AS and its clinical complications. NETs are characterized by three-dimensional web-like structures composed of a scaffold of decondensed chromatins decorated with modified histones, cytoplasmic, and granule proteins. 10 The majority of extracellular DNA originates from the nucleus, and the minority is from mitochondrial DNA. 11 A variety of proteins are identified in NETs and their changes depend on the stimulus, mainly including cytoplasmic and cytoskeletal proteins (e.g. calprotectin), metabolic enzymes in neutrophil granules (e.g. serine protease neutrophil elastase [NE], myeloperoxidase [MPO], and calprotectin), and antimicrobial peptides.12–14 Of note, NETs-derived extracellular DNA and protein components exert cytotoxic and proinflammatory effects, 15 possibly providing a causative link between inflammation and AS.
This review summarizes the current knowledge of the trigger mechanisms of NETs formation, and the contribution of NETs to AS and its complications. We also focused on the possibility of adopting NETs as promising biomarkers and potential therapeutic targets in clinical practice.
Mechanisms of NETs formation
The process of NETs release accompanied by programmed cell death is termed suicidal NETosis. In the light of differences of the pathogenic stimulations, membrane receptors, signaling cascades, and cytomembrane integrity, NETs are formed in at least two ways: suicidal NETosis and vital NETs, the latter including the release of nuclear DNA and mitochondrial DNA. 16 A schematic illustration of NETs types and the main regulatory mechanisms are shown in Figure 1.
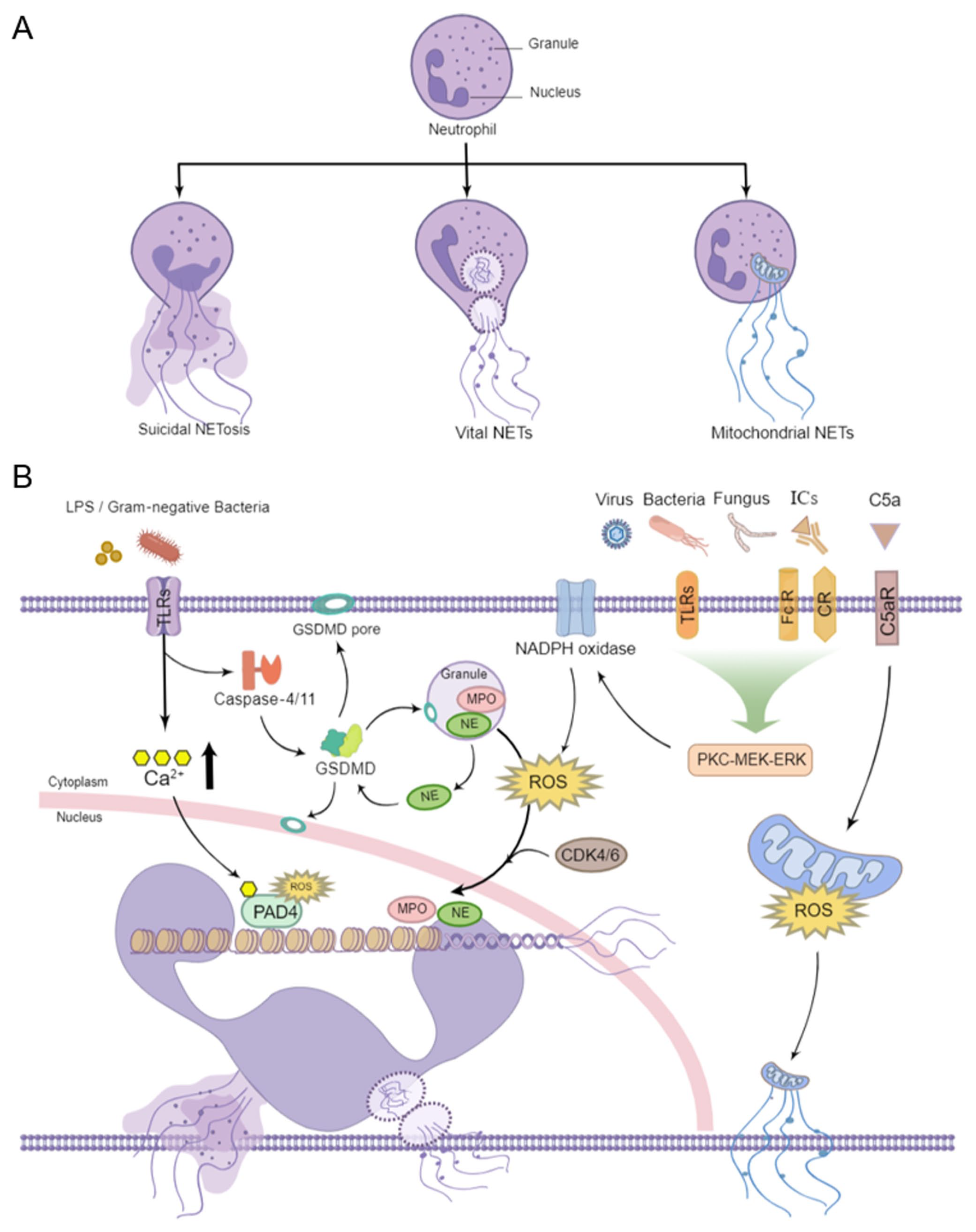
Schematic representation of types and regulatory mechanisms of NETs formation. (a) The classic suicidal NETosis is characterized by nuclear chromatin decondensation, membrane rupture, and cell death. The vital NETs occurred without destroying the cytoplasmic membranes, preserving the phagocytic function of neutrophils. The mitochondrial NETs contain mitochondrial DNA but not nuclear DNA. (b) Extracellular stimulation may result in ROS production through the activation of PKC–MEK–ERK via corresponding membrane receptors. Then, PAD4 is activated, and MPO and NE migrate to the nucleus, leading to chromatin depolymerization and suicidal NETosis. GSDMD and CDK4/6 are also essential regulators by promoting NE translocation to the nucleus during NETosis. The vital NETs occur as a rapid response to LPS and gram-negative bacteria with subsequent elevation of Ca2+ and direct activation of PAD4, resulting in the release of nuclear DNA. Following Ca5 stimulation, mitochondrial ROS is produced and the mitochondrial DNA is released instead of nuclear DNA, a process known as mitochondrial NETs.
Suicidal NETosis
During suicidal NETosis, neutrophils are activated with some stimuli,17,18 such as pathogenic microorganisms and immune complexes (ICs), through toll-like receptors (TLRs), antibody crystallizable fragment receptors (FcRs), and complement receptors (CRs). 19 Subsequently, the activation of downstream protein kinase C (PKC)/mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) cascade signaling pathway results in the assembly of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex and the production of reactive oxygen species (ROS), which triggers the translocation of MPO and NE from azurophilic granules to the nucleus facilitating the unfolding of chromatin. 20 In the meantime, protein-arginine deiminase type 4 (PAD4), a calcium-dependent nuclear enzyme, promotes histone citrullination, a process that contributes to chromatin decondensation. 21 Ultimately, the nuclear membrane is disrupted, chromatin decorated with granular and cytosolic proteins enters the cytoplasm, neutrophils initiate the cell death program, and then NETs are released. PAD4-mediated citrullination of histones is crucial in contributing to classical suicidal NETosis. 22 However, PAD4 inhibition by GSK484 failed to block DNA extrusion in the Entamoeba histolytica-triggered NETosis. 23 According to Claushuis et al., 24 both PAD4 +/+ and PAD4−/− mice showed similar levels of NET markers, cell-free DNA and nucleosomes, and organ injury during pneumonia-derived sepsis caused by Klebsiella pneumonia. These findings highlight the existence of additional pathways other than PAD4.
Gasdermin D (GSDMD) is a pore-forming protein that can cause pyroptosis, a form of inflammatory cell death via the activation of inflammasomes. Recent studies found that GSDMD can also promote the formation of NETs. The cleaved GSDMD liberates an amino (N)-terminal effector domain that incorporates into granule membrane, nuclear membrane, and cell membrane to form pores, and regulates the permeability of neutrophils, leading to the release of NETs into extracellular space.25,26 During NETosis, GSDMD is proteolytically activated by NE in a feedback loop and, in turn, promotes NE release from azurophilic granules and subsequent chromatin decondensation. 26 In addition, the cell-cycle regulator cyclin-dependent kinase (CDK) 4/6 is required for the duplication of centrosomes and also an essential regulator of NETs by promoting NE translocation to the nucleus. 27
Vital NETs
Unlike suicidal NETosis, vital NETs formation occurs as a rapid response to bacterial lipopolysaccharide (LPS) and gram-negative bacteria through TLR2/4. The subsequent increase in intracellular Ca2+ leads to the activation of PAD4 independent of NADPH oxidant, resulting in citrullination of histone H3 and H4 for chromatin decondensation. 16 Afterward, chromatin decorated with protein is expelled to the outside of neutrophils in the form of vesicles without nuclear or cell membrane disruption. 28 At this time, neutrophils are alive without nuclear DNA and still possess the ability to phagocytose and chemotaxis. 29 Although the NADPH oxidase–ROS axis is necessary for NETosis in most contexts, NADPH oxidase is not the only source of neutrophil ROS. Since mitochondria are one of the major sites of ROS generation, mitochondrial ROS production is sufficient to drive NETosis even in the absence of functional NADPH oxidase. 30 After pretreatment with granulocyte-macrophage colony-stimulating factor (GM-CSF) and subsequent stimulation with LPS or complement factor 5a (C5a), the released extracellular traps contain mitochondrial DNA rather than nuclear DNA. This process occurred in a mitochondrial ROS-dependent manner and retained the vitality of neutrophils. 31 The elevated mitochondrial DNA in NETs was also observed in patients suffering from systemic lupus erythematosus (SLE). 32
Contributions of NETs to AS
AS is a pathological process mainly caused by cholesterol deposition and inflammation dysregulation, involving various vascular and immune cells and influenced by multiple risk factors. 33 As an emerging mechanism, NETs were identified in the luminal locations in murine and human atherosclerotic lesions34,35 and arterial thrombosis,36,37 implying that NETs formation may occur at all stages of AS development. Also, in SLE patients with positive dsDNA antibodies, the enhanced NETosis was associated with proatherogenic dyslipidemia, accelerated AS, and increased cardiovascular risk. 38 Therefore, we describe the role of NETs based on the mechanism of AS formation, such as lipid metabolism, inflammatory immunity, intercellular communication, and risk factors.
Lipids
Lipid metabolism imbalance is generally considered to play a central role in the pathogenesis of atherosclerotic plaques. 39 The major sources of lipid accumulation in the arterial wall are proatherogenic modified low-density lipoprotein (mLDL), such as oxLDL, and monohydrate cholesterol crystals formed by excessive circulating cholesterol deposition, contributing to the progression of atherosclerotic plaques and even the final rupture.2,40
Neumann et al. 41 found primary human neutrophils demonstrated a significant release of NETs independent of NADPH oxidase when treated with different concentrations of methyl- when treated (Mhy– when treated with different concentrations Similar observations occurred in freshly isolated human neutrophils after stimulation with statins, inhibiting cholesterol metabolism. 42 In the context of AS, cholesterol crystals have been reported to trigger the release of NETs as sterile danger signals. 43 Unlike the influence of cholesterol content in the cell membranes, exogenous cholesterol crystals-stimulated NETs formation depends on NADPH oxidase–ROS axis and downstream translocation of NE to the nucleus. 43 And then, NETs prime macrophages for cytokine release, activating Th-17 cells that amplify immune cell recruitment in atherosclerotic plaques of the murine model. 43 As modified cholesterol, oxLDL induced NETs formation via TLR–PKC–Interleukin-1 receptor associated kinase (IRAK) - mitogen-activated protein kinase (MAPK) and NADPH oxidase activation in human neutrophils, 44 which might contribute to the development of AS. 45 In turn, the release of NETs and NETs-associated citrullinated histone that promote LDL oxidation, aggregation, and foam cell formation were found to be increased. 46
Chronic hyperlipidemia initiates the process of AS and maintains progressive lesions. 47 Neutrophils adhere to the lumen of large arteries during early stages of AS, and hypercholesterolemia induces an increase in the number of neutrophils. 48 NET structures were detected as early as three weeks in the aortic root atherosclerotic lesions after high-fat diet (HFD) feeding, became more abundant at four weeks, and continued to be present in the plaque after that. 49 The resolution of NETs by injecting DNase I into male Ldlr−/− mice fed with Western diet for 16 weeks suppressed NETs-induced AS progression in diabetic mice. 50 Also, using a photochemical injury model, hypercholesterolemic ApoE−/− mice were treated for 11 weeks with Cl-amidine to block NETs formation, which reduced atherosclerotic lesion area and delayed time to thrombosis. 51 Molinaro R et al. 52 developed collagen IV-targeted nanoparticles (Col IV NP) to deliver PAD4 inhibitor GSK484 selectively to regions with features of superficial erosion that resulted in decreased NETs accumulation at sites of intimal injury and preserved endothelial continuity in eight-week-old ApoE−/− mice. Furthermore, hypercholesterolemia impaired the NET-induced DNase response resulting in defective clearance and accumulation of NETs in the advanced atherosclerotic plaques. 53 These results shed light on the crucial role in the process from hypercholesterolemia to AS and thrombosis.
Inflammasomes
Inflammation is a crucial contributor and promising target of AS. It participates in all phases of AS from the onset to the development of complications through multiple mechanisms, such as inflammasomes,54,55 immune cells, and their secreted cytokines. 56 Inflammasomes are intracellular multiprotein complexes that comprise a sensor protein belonging to the absent in melanoma 2 (AIM2)-like receptor or the nucleotide-binding domain and leucine-rich repeat receptor (NLR) family, an adaptor protein of apoptosis-associated speck-like protein containing a caspase-activation and recruitment domain (CARD) (ASC), and an inactive zymogen, procaspase-1. 57 The assembled NLRP3 inflammasome can activate the protease caspase-1 to cleave pro-IL-1β into mature IL-1β in response to pathogen and damage-associated molecular patterns (PAMPs/DAMPs). 58 In Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) trial, antagonism of IL-1β reduced cardiovascular events in patients with a previous MI, pointing to a role of inflammasomes in atherothrombotic disease. 59 Another study showed that cholesterol efflux could decrease inflammasome activation via ATP-binding cassette transporters A1 and G1 (ABCA1/G1). 60 Inflammasomes appear to be a link between lipid metabolism and atherosclerosis as mediators of inflammation.
Aside from defective cholesterol efflux, uptake of cholesterol crystal and oxLDL can activate the inflammasome, while forming GSDMD-dependent pore in the plasma membrane of macrophages and neutrophils that may result in NETs release contributing to plaque erosion and thrombosis. 61 Furthermore, cholesterol accumulation in ABCA1/G1-deficient myeloid cells activates the NLRP3 inflammasome promoting neutrophil infiltration and NETs formation in mice atherosclerotic lesions, which points out the existence of cholesterol-NLRP3-NETs axis involved in the progression of AS. 62 Consistent with serving as a startup signal, the release of NETs desired for cholesterol crystals triggered the activation of NLRP3 inflammasome in macrophages with the mice atherosclerotic model. 43 The same study suggests a feedback loop that increased the synthesis of IL-1β from inflammasome in macrophages that, in turn, stimulate NETs formation in neutrophils. 43 As previously mentioned, GSDMD was cleaved into N-terminal fragments by activated caspase-11 to form pores in the neutrophil membrane and promote the release of NETs. 26 Meanwhile, GSDMD was also required in non-canonical NLRP3 inflammasome activation as the direct substrate of activated caspase-1. 63 Chen et al. 64 reported that neutrophil exposure to cytosolic LPS or cytosolic gram-negative bacteria (Salmonellae to cytosolic strate of activated caspase-1non-canonical (caspase-4/11) inflammasome signaling and triggered GSDMD-dependent neutrophil death and along with the release of NETs. Similarly, NETs-derived DNA induced by LPS in the lysosomes has been demonstrated to bind with the AIM2 sensor and activate the AIM2 inflammasome, causing alveolar macrophage pyroptosis. 65 Even though the relationship between the inflammasome and NETs is not entirely understood, recent discoveries of the GSDMD and AIM2 inflammasome may shed light on the relationship between these two in AS.
Crosstalk between NETs and other crucial cells residing in the plaque and circulation
The pathogenesis of AS can be summarized into several central stages focusing on the cell types involved: (1) ECs dysfunction as the initiation of AS, (2) monocytes enter the intima and differentiate to macrophages, and generate foam cells for the growth of the lesion, (3) vascular smooth muscle cells (VSMCs) migration for the advancement, and (4) platelet activation for thrombosis. NETs serve as a way of intercellular communication throughout the progression of the initiation and advancement of AS.
ECs
The vessel wall consists of a monolayer of ECs that becomes the first barrier to molecules, cells, or pathogens circulating in the blood flow. 66 As a result of disturbed blood flow, endothelial integrity is disrupted and becomes leaky, facilitating transendothelial transport or diffusion of plasma lipoproteins. 67 Then, ECs are activated to express adhesion molecules and chemotactic factors in response to the oxidation of lipoprotein lipids and other inflammatory mediators. 68 oxLDL Strongly accelerated NETs formation and the release of MPO and proteases from neutrophils, which act on native LDL to facilitate modification. The coexistence of NETs and oxLDL or native LDL contributed to endothelial inflammation to cause cell elongation and enhanced MMP-1 production in ECs. 69 In another study, oxLDL-treated human umbilical vein endothelial cells (HUVECs)-derived exosomes were demonstrated to trigger hyperlipidemia, inflammatory response, and NETs release in a mouse model of AS. 70 Moreover, the release of NETs promoted vascular leakage and endothelial-to-mesenchymal transition through the degradation of VE-cadherin and the activation of ofcatenin signaling. 71 In addition to dyslipidemia-induced NETs, hyperglycemia has been proven to enhance NETs generation in a NAPDH-dependent pathway, 72 eventually contributing to the damage of endothelial glycocalyx. 73 In a diet-induced obesity mouse model, the inhibition or degradation of NETs could reduce the endothelial dysfunction. 74 However, disturbed flow triggers the TLR2-dependent ECs apoptosis and secretion of IL-8 leading to the release of NETs, which, in turn, aggravates the injury of the endothelial layer and enhances the thrombus formation in plaque erosion. 75
Monocytes and macrophages
Monocytes are recruited to the endothelial layer and migrate into the intima in response to the expression adhesion molecules and chemotactic factors expressed by ECs. They are differentiated into macrophages and uptake-modified lipoproteins, yielding foam cells that frequently undergo apoptosis or necrosis to give rise to necrotic core in the lesion. 76 Of importance is macrophages phenotypic plasticity in response to microenvironment involving in atherosclerotic lesion size, composition, and stability. 77 In a hypercholesterolemic mice model, a single dose of LPS injection was able to expand the size of the atherosclerotic lesions via the stimulation of NETs release in the arterial lumen. 78 In the same study, NETs-resident histone H2a attracted monocytes in a receptor-independent, surface charge-dependent fashion accelerating AS. The release of NETs triggered by cholesterol crystals primed macrophages for cytokine release, activating Th-17 cells that amplify immune cell recruitment in atherosclerotic plaques. 43 Alterations of the microenvironment within atherosclerotic plaques affect the crosstalk between NETs and macrophages. Under normoxia, NETs significantly increased the expression of inflammatory markers and uptake of oxLDL in M1 macrophages, but not under hypoxic conditions. 79 In turn, oxLDL exosomes released from oxLDL-stimulated macrophages, enriched with miR-146a, induced NETs to augment atherosclerotic lesion formation. 80 Also NETs, effectively induced by a nicotine administration to the HL-60 cell-derived neutrophil-like cells, promoted atherogenesis via suppressing 7-ketocholesterol mediated by both autophagosome formation and autophagosome–lysosome fusion in macrophages. 81 Haider et al. 82 showed that macrophages can degrade NETs through an interplay of extracellular and secreted DNases followed by intracellular uptake, and this effect was boosted by proinflammatory polarization of macrophages via micropinocytosis, leading to thrombus resolution in vivo. These evidences suggest that a counterbalance of NETs generation and degradation is necessary to maintain appropriate homeostasis.
Smooth Muscle Cells
In response to injury, SMCs transform to a proliferative activity, migrate into the intima, and increase the production of extracellular matrix (ECM) components contributing to fibrous cap. Recent studies have discovered that SMCs can undergo transdifferentiation into macrophage-like cells that take up lipid into foam cells according to single-cell RNA sequencing.83,84 Thus, SMCs are involved in two major processes of the destabilized plaques, characterized by necrotic core growth and fibrous cap thinning. The physical interaction between resident SMCs and neutrophils in the fibrous cap is able to induce NETs formation by Ced plaques, characteriznd 7 (CCL7) synthesized from activated SMCs. 85 Among the nuclear proteins released from NETs, histone H4, in turn, binds to and lyses SMCs, leading to the destabilization of plaques. Consistent with this, human neutropenia increased SMC content and fibrous cap thickness, which resulted in lower overall plaque vulnerability. 86 However, NETs promote SMCs proliferation via Akt/CDK inhibitor 1b (CDKN1b)/ thymidine kinase 1 (TK1) in spontaneously hypertensive rats. 87 These findings suggest that changes in the number of SMCs within plaques seem to associate with increased SMCs death, but not with changes in cell proliferation. 86 It is unclear whether NETs are involved in the SMCs phenotypic transition to macrophage-like SMCs and the formation of smooth muscle foam cells, and further investigation is still needed.
Platelets
Platelets are the key mediator of plaque rupture and atherothrombosis, and yet there is growing evidence that they play an inflammatory role as an immune cell and participate in the development of AS. 88 When the vascular microenvironment changes, platelets circulating in the blood can be activated rapidly to participate in AS, through adhering to damaged blood vessels to maintain blood vessel integrity during endothelial dysfunction and recruit immune cells to promote the transmigration across the intima. 89 In early atherogenesis, platelet-derived CCL5 and CXCL4 interact to form heterodimers, then synergistically recruit monocytes to inflammatory vascular ECs 90 and induce NETosis formation, 91 providing a link to NETs in the luminal aspects with progressive atherosclerotic lesions. 34 Afterward, a self-sustaining loop of reciprocal platelet and neutrophil activation is initiated by the prothrombotic microenvironment. 92 The loop includes activated neutrophils secrete cathelicidins, LL-37, or cathelicidin-related antimicrobial peptide (CRAMP), promoting further platelet activation and aggregation, 93 and activated platelets secrete HMGB1 promoting NET formation, thereby sustaining platelet activation and thrombosis stability.37,94 Mechanistically, integrin ntegrin tically, integrin "rete HMGB1 promoting NET formation, thereby sustaining platelet activati 95 And complement C3 mediates NETs-induced platelet activation and aggregation accelerating thrombosis.96,97 Furthermore, Hally et al. 98 combining biomarkers of NETosis and platelet activation were independent predictors of one-year major adverse cardiac events (MACE) in patients with acute myocardial infarction (AMI).
Aging
AS is a disease of both organ aging and cellular senescence, and thus, increasing age is a major risk factor for atherosclerotic CVD. 99 Regarding the contribution to AS about aging-regulated NETs formation, two potential mechanisms are discussed. First, aging can result in the acquisition of hematopoietic stem cells (HSCs) with somatic mutations in certain genes, such as TET2, DMMT3A, and JAK2, leading to the clonal expansion of proatherogenic myeloid cells in peripheral blood. 100 This process is known as clonal hematopoiesis of indeterminate potential (CHIP) associated with the risk of adverse cardiovascular outcomes. 101 In hypercholesterolemic mice, hematopoietic JAK2V617F expression promoted early lesion formation with neutrophil infiltration and increased complexity in advanced AS with cellular defects in erythrocytes and macrophages. 102 Moreover, increased propensity for NET formation and thrombosis was observed in mice with conditional knock-in of Jak2V617F, mimicking a clonal hematopoietic disease of myeloproliferative neoplasms (MPNs). 103 Second, chronic low-grade inflammation developed with aging, characterized by aberrant activation of the immune cells, enhanced expression of proinflammatory cytokines, such as IL-1β, and activation of ICs, such as the NLRP3 inflammasome, is now being recognized as a key driver of aging-related pathologies, such as AS. 104 In light of the earlier revelation that NLRP3 inflammasome and IL-1β were involved in NETs formation, it seems the aging-inflammation-NETs-AS linkage is quite plausible. NETs were significantly increased in the atherosclerotic lesions of aged mice compared with lesions of younger mice. In this study, mitochondrial catalase (mCAT) transgenic mice were used for ectopically expressing human catalase gene in mitochondria, which reduced mitochondrial oxidative stress (mitoOS). The aged Ldlr−/− mice transfected with mCAT had smaller lesions and decreased NETs compared with age-matched controls, similar to the character in young mice. 105 Interestingly, aged neutrophils themselves in the circulation represent an overactive subset exhibiting enhanced NETs formation under inflammatory conditions,106,107 which is a possible contributor to inflamm-aging, accelerating AS and thrombosis and promoting plaque rupture and erosion. 108
NETs as biomarkers in AS and AS-associated disease
Since NETs are implicated in the pathological mechanisms of AS, their components may be promising biomarkers of the AS severity and clinical outcomes of AS-associated disease in terms of diagnosis or prognosis. The circulating levels of double-stranded DNA, MPO–DNA complexes, guanylated histones, and NE are often used as a way of assessing NETs. In patients with suspected or stable coronary artery disease, the elevated dsDNA and MPO–DNA complexes in plasma were positively associated with the severity and the number of atherosclerotic vessels, and the occurrence of MACE.109,110 Patients with acute coronary syndromes (ACS) at initial presentation have elevated circulating levels of NETs. 111 In a cohort of 259 patients with ST-segment elevation myocardial infarction (STEMI), dsDNA levels on high circulating day 1 after percutaneous coronary intervention (PCI) were associated with myocardial infarct size, more adverse left ventricular remodeling, poor clinical outcome during long-term follow-up, 112 and even increased risk of death. 113 Combined scores of biomarkers for NETs formation and platelet activation were independent predictors of MACE within one year following AMI. 98 Not only in cardiovascular areas, NETs-forming neutrophils were also found throughout brain tissue of ischemic stroke patients, and elevated plasma NETs-related biomarkers correlated with acute ischemic stroke (AIS) of first occurrence and also worse outcomes.111,114
Neutrophils are one of the most abundant cell subsets that accumulated in arterial thrombi with patients suffering from AMI, and pharmacological inhibition of NETs formation reduced arterial thrombosis and limited injury in a mouse model. 115 Concordant with this result, NETs-activated platelets destroyed the endothelial barrier and induced the procoagulant activity, contributing to thrombogenicity in carotid stenosis and AIS. 116 In addition, NET-derived components also relate to atherosclerotic and thrombotic aspects of other diseases. 117 For example, in maintenance of hemodialysis patients, uremia-associated-increased NET formation was correlated with an increased burden of AS. A prospective cohort study conducted in 38 cancer-related stroke patients, 27 patients of active cancer but no stroke, 40 patients of AIS but no cancer, and 33 healthy controls suggested increased circulating DNA levels were associated with cancer-related stroke. 118 In patients with COVID-19, the levels of NET-derived components in both circulation and thrombotic lesions were relevant to higher risk of morbid thrombotic events in spite of prophylactic anticoagulation.119,120
NETs-targeted therapeutic strategies
In view of the importance associated with lipid metabolism, inflammation, and AS, NETs are promising as an emerging therapeutic target with regard to NET components and release pathways. PAD4 is an essential enzyme that converts arginine into citrulline for chromatin decondensation in NETs formation. In the PAD4-deficient mice either genetically or pharmacologically, NET formation and the expression of inflammatory factors were inhibited and the plaque area decreased in the aortic region.49,51 Moreover, PAD4 deficiency in bone marrow-derived cells or administration of DNase I to disrupt NETs decreased the extent of arterial intimal injury and thrombosis formation in LDL gene knockout mice. 121 Cl-amidine targets several isoforms of PAD with off-target effects and hence lacks the therapeutic specificity as a first-generation PAD inhibitor. Recently, the novel and selective PAD4 inhibitors, GSK199 and GSK484, were validated the critical role of human and mouse PAD4 in both histone citrullination and NETs formation through binding to a calcium-deficient form. 122 GSK484 can reduce NET accumulation at sites of intimal injury and preserved endothelial continuity 52 and protect cardiomyocytes from MI-induced NET formation and inflammatory cytokine secretion, in turn, alleviating cardiac ischemia-induced apoptosis of cardiomyocytes. 123 In GSK199-treated mice, platelet high-mobility group box-1 (HMGB1)-mediated NET formation was reduced, thus reducing deep venous thrombosis (DVT). 124 In addition, a therapeutic anticitrullinated protein antibody (tAPCA) can inhibit NET release and improves signs of inflammation in a mouse model of arthritis via specifically binding to citrulline at position 3 (Cit3) in histone 2a (citH2a) and 4 (citH4). 125 Studies with these inhibitors of PAD4 could provide an approach to limiting NET formation, which maybe also the clinical therapeutic strategies for AS.
Beyond limiting NETs release, dissolving NETs may be another beneficial approach in NET-mediated AS and thrombosis. In atherosclerotic mice model, DNase I administration reduced NETs formation, lesion size, 43 and decreased superficial erosion of the endothelium. 121 Dornase alfa, a recombinant human DNAase I for digesting the DNA backbone of NETs, is already in a phase II clinical trial (NCT04785066) for the treatment of ischemic stroke. Vogel et al. 126 and Ge et al. 127 observed DNase1 accumulation in the infarcted region of the heart and effective disruption of extracellular cytotoxic chromatin and subsequent reduction of high local histone concentrations for protecting myocytes. The disruption of NETs by DNase I ameliorated thrombolytic therapy for ischemic stroke by reducing tissue plasminogen activator (tPA)-associated hemorrhage. 128
With a significant role in NETs release, GSDMD is cleaved by neutrophil proteases and, in turn, affects protease activation and nuclear expansion forming a feed-forward loop. 26 Compared with chronic inflammatory diseases, such as atherosclerosis, inhibiting GSDMD may be a more effective treatment in acute inflammatory injury. Silva et al. 129 demonstrated that inhibition of GSDMD with disulfiram or genic deletion abrogated NETs formation, reducing multiple organ dysfunction and sepsis lethality. In a mouse model of SARS-CoV-2 infection, the treatment with GSDMD inhibitor reduced NETs release and organ damage. 130 Beyond that, neutralization of NETs-related proteins may also be a potential treatment for atherosclerotic inflammation. Therapeutic interference of histone H2a with antibodies or silico designed cyclical peptides reduced luminal monocyte adhesion and plaque expansion promoted by NETs. 78 A similar approach was used for the neutralization of histone H4 that prevented cell death of SMCs and stabilizes atherosclerotic lesions. 86
Conclusions
Although neutrophils have long been recognized in response to acute inflammation, emerging evidence revealed their role in chronic inflammation since the discovery of NETs, as atherosclerosis. As the link between lipid metabolism, inflammation, immunity, and atherogenesis, NETs are involved in various stages of the AS pathogenesis, and thus used as a promising biomarker for the diagnosis or prognosis and a potential therapeutic target in AS-related diseases. However, much work remains to be done in the development, standardization, and validation of NETs as a biomarker before it can be adopted into clinical practice. It is important to keep in mind that the capability of neutrophils to produce NETs and the actions performed by NETs in the context may be determined by environmental conditions. Further research efforts and clinical evaluation are needed in terms of therapeutic aspects, such as more specific targeting modalities and less occurrence of side effects. Despite these challenges, new insights from the NETs study are promising to provide new breakthroughs in improving the diagnosis and treatment of AS and its related diseases.
Footnotes
Authors’ Contributions
CG, BP, and GL had the idea for the article. SS and CA designed the article framework. MW, NW, and YY performed the literature search. The work was drafted by CG and critically revised by GL. All authors commented on previous versions of the article, and read and approved the final article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (81803975).