
Abstract
This review summarizes the current knowledge of fibroblast growth factor 23 signaling in bone and its role in the disease pathology of X-linked hypophosphatemia. Craniosynostosis is an under-recognized complication of X-linked hypophosphatemia. The clinical implications and potential cellular mechanisms invoked by increased fibroblast growth factor 23 signaling causing craniosynostosis are reviewed. Knowledge gaps are identified and provide direction for future clinical and basic science studies.
Keywords
Impact Statement
Craniosynostosis occurs commonly in patients with X-linked hypophosphatemic (XLH) rickets. We review and highlight what is known and not known about this phenomenon including exploring possible molecular mechanisms.
Introduction: XLH and the role of PHEX and FGF23 on bone health
X-linked hypophosphatemia (XLH; OMIM 307800) is a renal phosphate-wasting disorder that is the most common heritable form of rickets, affecting approximately 1 in 20,000 individuals. 1 XLH is caused by inactivating pathogenic variants in the phosphate-regulating endopeptidase homolog X-linked (PHEX) gene. 2 Primarily expressed in osteoblasts, osteocytes, and odontoblasts, PHEX inactivation leads to increased production of fibroblast growth factor 23 (FGF23) by osteocytes.3–7 PHEX also regulates the levels of potent bone mineralization inhibitors, including osteopontin (OPN) and matrix extracellular phosphoglycoprotein (MEPE).3,8 Fibroblast growth factor 23 (FGF23) is released into the circulation and binds the Klotho–fibroblast growth factor receptor 1 (FGFR1) complex in the renal proximal tubule which induces phosphaturia and hypophosphatemia.9,10 FGF23 excess and resulting hypophosphatemia have well-known clinical sequelae, including rickets and osteomalacia, short stature, lower limb deformity, muscle weakness, bone pain, dental abscesses, and insufficiency fractures; enthesopathy and spinal stenosis are additional comorbidities in adults with XLH. 11 Craniosynostosis is seen in up to 59% of PHEX-positive individuals, with the sagittal suture most commonly affected.12,13 The Hyp mouse, a murine model of XLH harboring a loss-of-function variant in the PHEX gene, also demonstrates craniofacial and cranial suture abnormalities, and has been a useful model in exploring the cellular pathology in XLH. However, the mechanisms mediating craniosynostosis in XLH have not been completely elucidated.2,14
FGF23 regulation and post-translational modifications
FGF23 is synthesized as a 251 amino acid pro-protein that undergoes post-translational modification to regulate its activity by cleavage of 24 amino acids to form active FGF23. 15 FGF23 contains a 176RXXR179 motif that is integral for FGF23 inactivation since active FGF23 is cleaved at this site, releasing C and N terminal fragments. The released C terminal fragment may further suppress FGF23 action, as the C terminus can bind Klotho–FGFR1 and impair FGF23 signaling, acting as a negative inhibitor of FGF23 activity. 16 FGF23 inactivation is dependent on the activity of Family with Sequence Similarity 20, member C (FAM20C) and Furin. 17 FAM20C is a protein kinase that phosphorylates the serine-180 residue immediately adjacent to the 176RXXR179 motif. Furin, a protease, then cleaves phosphorylated FGF23 into its N and C terminus. FGF23 pathogenic variants at the 176RXXR179 motif cause Autosomal Dominant Hypophosphatemic Rickets (ADHR, OMIM 193100), as variants in this proteolytic recognition sequence render FGF23 resistant to cleavage and thus lead to persistent biological activity. 18 Therefore, the 176RXXR179 motif is essential for post-translational modification and regulation of FGF23 activity. FGF23 undergoes O-glycosylation at this motif which protects FGF23 from degradation. 19 In the absence of O-glycosylation, FGF23 undergoes excessive cleavage and the loss of full-length, biologically active FGF23 results in familial tumoral calcinosis. 20
FGF23 effects on calcium/phosphate homeostasis
FGF23 is a phosphaturic hormone that causes renal phosphate-wasting; the suppressive effect of FGF23 excess on 1,25-dihydroxyvitamin D synthesis also impairs gastrointestinal phosphate absorption. FGF23 is primarily secreted by osteoblasts and osteocytes; 21 other sources of FGF23 include odontoblasts. Both phosphate and calcitriol 1,25-dihydroxyvitamin D regulate FGF23 levels, although phosphate is likely the more critical determinant of FGF23 secretion.22,23 It has recently been demonstrated that the kidney detects phosphate levels through glycolysis, resulting in increased FGF23 production. 24 FGF23 decreases expression of the type IIa and IIc sodium–phosphate cotransporters in the kidney proximal tubule which results in increased renal phosphate excretion.10,25,26 FGF23 both inhibits the formation of 1,25-dihydroxyvitamin D and increases inactivation of 25-hydroxyvitamin D. FGF23 inhibits renal 1-alpha-hydroxylase expression, impairing the conversion of 25-hydroxyvitamin D to its active form, 1,25-dihydroxyvitamin D.25,26 Furthermore, FGF23 increases 24-hydroxylase activity to convert both 1,25-dihydroxyvitamin D and 25-hydroxyvitamin D to their inactive 24-hydroxylated forms.27,28 Together, the net effect is renal phosphate-wasting along with impaired gastrointestinal phosphate and calcium absorption, contributing to the hypophosphatemia seen in disorders with increased FGF23. Inappropriately, normal or low 1,25-dihydroxyvitamin D in association with hypophosphatemia is considered a hallmark of FGF23-mediated renal phosphate-wasting. This is in contrast to non-FGF23-mediated hypophosphatemia, where 1,25-dihydroxyvitamin D is appropriately high in response to low serum phosphate levels, resulting in hypercalciuria (as for example, hereditary hypophosphatemia with hypercalciuria, a disorder resulting from loss of renal sodium–phosphate co-transport, but low serum FGF23 levels). 29
FGF23 excess causes multiple diseases, including XLH, autosomal dominant hypophosphatemic rickets, fibrous dysplasia-related hypophosphatemia, linear sebaceous nevi syndrome (cutaneous-skeletal hypophosphatemia syndrome), and tumor-induced osteomalacia.30,31 XLH is the prototype of FGF23-mediated hypophosphatemia, characterized by renal phosphate-wasting and typically inappropriately low/normal 1,25-dihydroxyvitamin D. 11 This can be quantified by calculating the tubular maximal resorption of phosphate per glomerular filtration rate (TmP/GFR = plasma phosphate−[urine phosphate/urine creatine × plasma creatinine]) with comparison to age- and sex-based normative ranges.32–35 Phosphate is a necessary component of hydroxyapatite along with calcium; as such, hypophosphatemia results in osteomalacia at the bone tissue level, defined in histomorphometric terms as an increase in osteoid thickness associated with prolongation of the mineralization lag time. Clinical consequences of osteomalacia include bowing deformity, bone pain, and “looser zones,” also known as insufficiency fractures or “pseudofractures.” The latter term is falling somewhat out of favor since the bone is indeed cracked in osteomalacia-induced looser zones and is correspondingly painful. Hypophosphatemia also interferes with mineralization of the growth plate, resulting in rickets which is apparent clinically as bowing deformity and poor growth, and radiographically typical features that include widening, fraying, and splaying of the growth plate.
PHEX and ASARM peptides: inhibition of mineralization
PHEX is an endopeptidase that was originally thought to degrade FGF23 directly. 36 However, it has been demonstrated that PHEX does not directly cause FGF23 degradation but rather PHEX degrades the mineralization inhibitors (“minhibins”) OPN and MEPE, with PHEX loss of function resulting in increased minhibins.3,8 Both OPN and MEPE contain acidic-serine-aspartate-rich motif (ASARM) proteolytic peptides which are responsible for the minhibin activity. ASARMs bind to hydroxyapatite to impair crystal formation and augment osteoclast adhesion to bone, promoting bone resorption.21,37–42 ASARMs also inhibit mineralization around the osteocyte to create the classic peri-osteocytic lesions unique to XLH. 43 Mineralization defects are similarly seen in Hyp mice where PHEX loss of function results in high serum and bone tissue levels of MEPE, OPN, and ASARM peptides, and osteomalacia, which is observed as seams of osteoid adjacent to mineralized matrix regions on histology.3,44,45 ASARMs also feedback to inhibit PHEX activity and increase FGF23 expression. 8 FGF23 is then released into the circulation where it promotes renal phosphate-wasting leading to systemic hypophosphatemia and reduced phosphate availability for hydroxyapatite formation. 8 Thus, the PHEX-OPN-ASARM-FGF23 pathway contributes to local peri-osteocytic mineralization defects and systemic hypophosphatemia and osteomalacia (Figure 1).
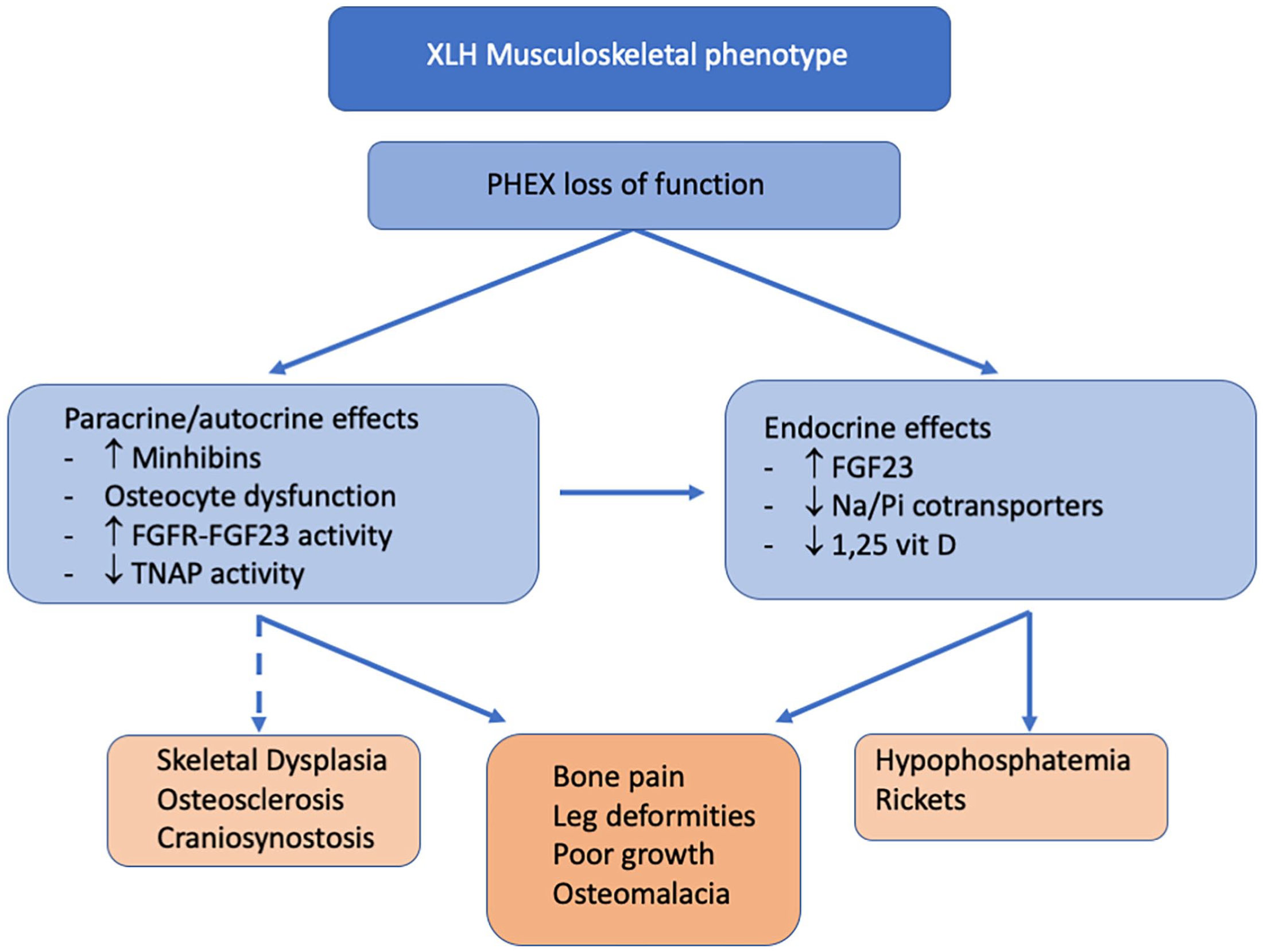
The impact of PHEX loss of function on bone mineralization.
XLH and craniosynostosis
Craniosynostosis is the premature fusion of cranial sutures which can occur in isolation or as part of some genetic syndromes. 46 Craniosynostosis occurs with a high frequency in XLH and the sagittal suture is most commonly affected (Figure 2), although there have been reports of multiple other suture involvement, including coronal, lambdoid, metopic, and pansynostosis.12,13,47,48 Premature fusion of the sagittal suture leads to a long and narrow (dolichocephalic) skull shape. This may also contribute to the classic skull phenotype of XLH with frontal bossing, posterior protuberance, and lateral narrowing. 15 The cranial index is a ratio of skull width to length, and a cranial index of 75% or less indicates skull deformity. It would be ideal if a simple external measurement such as this could diagnose craniosynostosis in these patients. However, some individuals with XLH and craniosynostosis have a normal cranial index. Consistent with this, several reports have found a normal cranial index and absence of dolichocephaly in children with XLH despite known synostosis.12,13 It was postulated therefore that these children developed craniosynostosis later in childhood, once the majority of skull growth was complete. 12 Other reports have demonstrated craniosynostosis presenting early in infancy, evident in a case series of two infants presenting by three months of age, before they were diagnosed with XLH, and before hypophosphatemia or rickets was apparent. 48 Both these children had evidence of skull deformity including dolichocephaly, frontal bossing, and endocortical scalloping. 48 A phenotype–genotype correlation does not exist for any of the clinical features in XLH, although truncating mutations are predicted to cause more severe disease.49,50 A phenotype–genotype correlation has not been demonstrated for craniosynostosis in patients with XLH. The average age at which craniosynostosis is evident clinically is presently unknown, nor whether the condition has its origin in utero or whether it tends to develop later in childhood. Prospective studies are necessary to determine the prevalence and natural history of craniosynostosis in XLH.
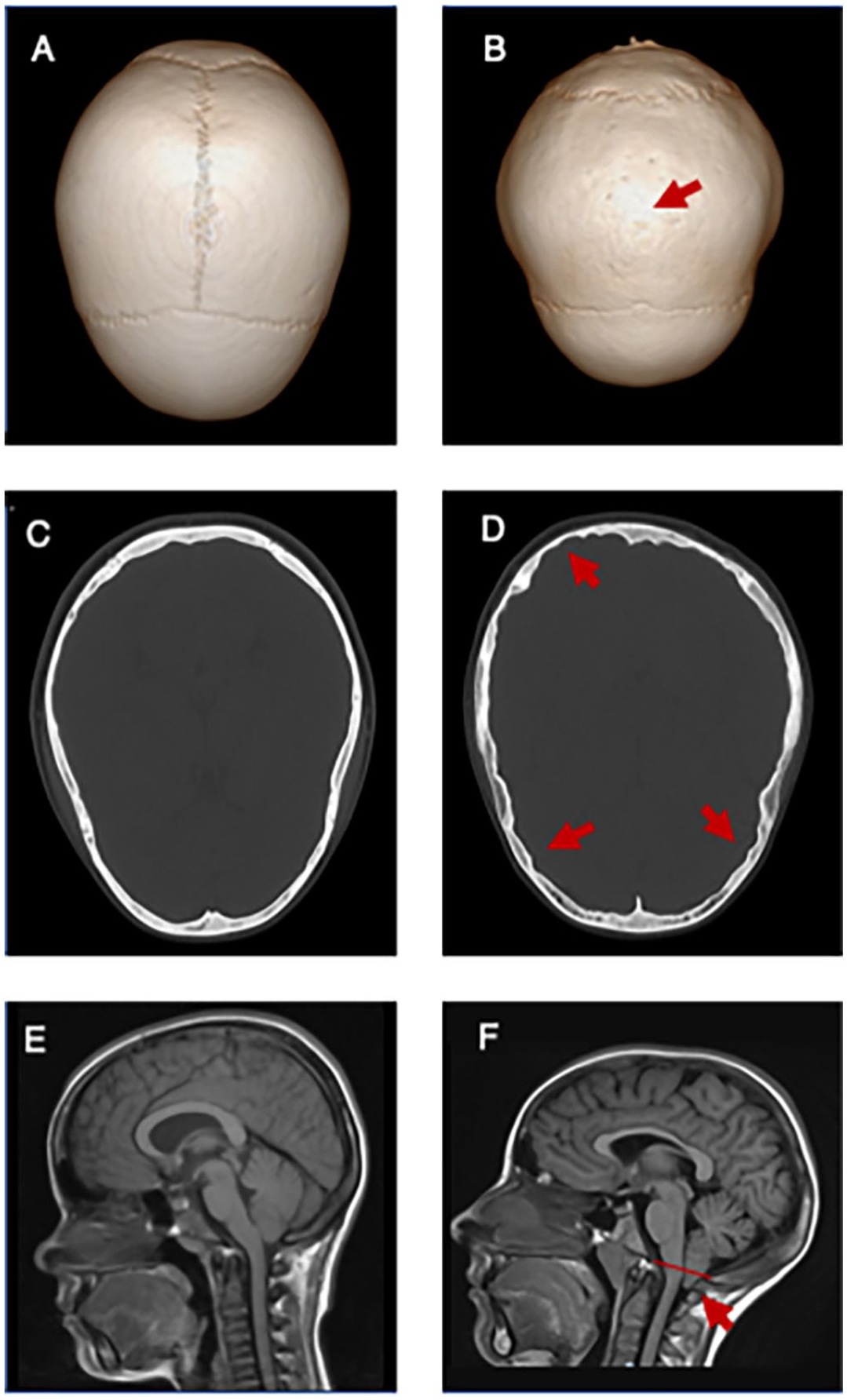
Craniosynostosis, endocortical scalloping, and Chiari 1 malformation (CM1) in XLH patients. (A/C/E) Normal cranial imaging in a 16-year-old female with XLH. Skull CT demonstrated normal sutures (A) and no endocortical scalloping (C). (E) Brain MRI demonstrated a normal craniocervical junction without a CM1. (B/D/E) Abnormal cranial imaging in individuals with XLH. (B) Sagittal synostosis in a 15-year-old male with XLH. Note the absence of the midline suture (red arrow) with lateral bulging of the skull and shortening of the anteroposterior axis. (D) Head CT in an 11-year-old male with XLH demonstrating endocortical scalloping. Note the ruffled appearance of the interior skull border (red arrows). (F) Brain MRI in a 13-year-old female with XLH and CM1. There is a 7-mm tonsillar ectopia (red arrow) through the foramen magnum (red line).
Multiple case series and retrospective reviews have reported their experience with craniosynostosis in XLH, detailing symptoms, and the need for surgical intervention.13,47,51–54 There are frequent reports of craniosynostosis, increased intracranial pressure, papilledema, and endocortical scalloping, along with central nervous system (CNS) abnormalities, such as Chiari 1 malformation (CM1) and syringomyelia. Figure 2 demonstrates craniosynostosis, endocortical scalloping, and CM1 in XLH. These neurologic complications can be severe requiring urgent neurosurgical intervention and cranial vault remodeling if not detected in a timely fashion.12,47,51,55 Fortunately, the surgical outcomes have been positive with the normalization of head shape, resolution of ophthalmologic and neurologic symptoms, and no reports of mortality (recognizing that the latter point may reflect publication bias against unfavorable outcomes). The cellular mechanisms are not well understood, although there are signaling abnormalities in XLH that may contribute to the development of craniosynostosis, such as altered FGFR signaling, altered tissue non-specific alkaline phosphatase (TNALP) activity, or altered heparan sulfate signaling. The potential cellular mechanisms are explored below.
Possible complications of craniosynostosis: Chiari 1 malformations
Chiari 1 malformations are craniovertebral abnormalities that are more common in XLH, where the cerebellar tonsils protrude out of the foramen magnum into the cervical spinal junction. Craniosynostosis has been hypothesized to contribute to CM1 in XLH, as inappropriate suture fusion restricts brain growth leading to downward pressure and protrusion of the cerebellar tonsils. The largest study to date reported CM1 in 25% of children with XLH, of which 9% required neurosurgical intervention. 13 Most of these children also had craniosynostosis, although one child had CM1 without craniosynostosis. CM1 may impair cerebrospinal fluid flow as the cerebellum limits flow through the foramen magnum. This can result in syringomyelia and cysts around the spinal cord. There are case reports of syringomyelia in XLH but the prevalence remains unknown. CM1 and syringomyelia have been associated with cervical and cranial neuropathy including bulbar palsy, papilledema, and hydrocephalus, and non-specific symptoms, including headaches and neck pain.13,54,56 It is increasingly recognized that XLH has higher rates of CNS anomalies. As such, clinicians should monitor for neurologic symptoms and pursue diagnostic imaging. At our centers, we carry out routine CNS imaging during the pediatric years even in the absence of symptoms to detect craniocervical abnormalities that require close monitoring as the child progresses through adolescence and adulthood.
XLH: a contradiction of osteomalacia and osteosclerosis
There is a phenotype of skull calvarium thickening that has been reported in XLH. In a description of two infants presenting with craniosynostosis, skull radiographs showed diffuse calvarial thickening in one of the patients. 48 Other reports have commented on areas of thickened calvarium interspersed with areas of soft thinned bone.47,48,51 XLH is known to cause an osteosclerotic phenotype despite the mechanical weakness of the bone.57,58 It is postulated that osteocytes sense the mechanical instability that underlies XLH and respond with increased trabeculae and bone formation. 59 However, the underlying bone remains soft and poorly mineralized, further contributing to the pathology. It is hypothesized that in addition to craniosynostosis, excessive bone formation and thickening of the skull may contribute to decreased intracranial space and the CM1. In addition to decreased intracranial space contributing to the CM1, it may also contribute to endocortical scalloping resulting in radiographic features referred to as a “copper-beaten skull” or “thumbprinting.” Together, these observations support that XLH is a skeletal dysplasia with inherent abnormal bone formation, rather than an isolated mineralization disorder.
Cellular basis of craniosynostosis in XLH
The cranial sutures are mechanosensitive spaces between two opposing cranial bones that facilitate appositional growth of the cranial bones. This process comprises undifferentiated proliferating osteogenic stem cells, some of which will differentiate into osteoblasts to form new bone along the skull edge. 46 Loss of this undifferentiated suture space causes craniosynostosis. 60 The Hyp mouse has craniofacial abnormalities that are similar to individuals with XLH including a shortened and narrow skull, frontal bossing, and synostosis (although the coronal suture is typically affected in the Hyp mice, compared with the sagittal suture in humans with XLH). Histologically, the sutures of Hyp mice show abnormalities as early as three weeks of age (i.e. at the time of weaning) with oblique collagen fiber bundles and fibroblasts that traverse the sutures. This differs significantly from wildtype mice where collagen fibers and fibroblasts orient parallel to suture surfaces; these differences indicate an altered mechanical environment in the disease state, in addition to reduced bone formation.61–63 Despite the high frequency of craniosynostosis in XLH, the underlying mechanism is incompletely understood. FGFR is present in the cranial suture and the majority of syndromic synostosis is due to variants in FGFR1/2/3. 46 FGF23 can bind to FGFR2/3 and it has been postulated that FGF23–FGFR2/3 binding at the cranial suture contributes to craniosynostosis, like other conditions with activated FGFR2/3. 64 Thus, excess FGF23 may impair bone apposition at the suture line, mediate craniosynostosis through FGFR2/3 binding and skull osteomalacia.
Role of FGFR in craniosynostosis
FGFR activation may play an important role in FGF23 signaling and formation of craniosynostosis. Hyp mice have increased Fgfr1 and Fgfr2 expression, which is present before birth. This suggests that FGF/FGFR signaling may be present in utero and persists postnatally. 65 Craniosynostosis is also seen in many genetic syndromes, such as Apert (OMIM 101200), Crouzon (OMIM 123500), Pfeiffer (OMIM 101600), Muenke (OMIM 602849), and Antley–Bixler syndrome (OMIM 207410). These conditions are associated with pathogenic variants in FGFR1, FGFR2, and FGFR3 with FGF23 mediating effects through each of these receptors.15,46 Hence, altered FGF signaling appears to be a common mechanism linked to craniosynostosis. The deletion of Fgfr1 in Hyp mice partially rescued the phenotype. These animals display decreased expression of fgf23 in bone, lower serum FGF23, improved phosphate and 1,25-dihydroxyvitamin D levels, and increased femur length and cortical thickness. 66 Fgfr1 deletion in osteocytes caused reduced gene expression of MEPE and FGF23, demonstrating that FGFR1 activation plays a role in regulating minhibins. 66 Thus, FGFR signaling abnormalities may mediate craniosynostosis in XLH by increasing both minhibins and FGF23. This also speaks to the possibility that systemic FGF23 might directly affect appositional bone growth and bone formation at the sutures.
TNALP and craniosynostosis
TNALP may play an important role in the development of craniosynostosis. TNALP is necessary to convert pyrophosphate (PPi) into inorganic phosphate (Pi), a key component in hydroxyapatite formation and bone mineralization. 67 In the Hyp mouse model of XLH, elevated FGF23 suppresses TNALP expression and causes an accumulation of PPi. Blocking FGF23–FGFR3 signaling helps restore normal TNALP expression and decreases PPi, permitting mineralization of bone matrix. 4 Furthermore, the incidence of craniosynostosis in TNALP-deficient mice correlates with circulating FGF23 levels. 68 Impaired TNALP activity or expression is seen in craniosynostosis conditions, such as hypophosphatasia and Crouzon syndrome. Hypophosphatasia is due to a loss of function of the ALPL gene, resulting in significantly reduced TNALP activity (with infantile hypophosphatasia being characterized by the presence of craniosynostosis). 69 The sagittal suture is typically involved in hypophosphatasia, similar to XLH, although hypophosphatasia has higher reported rates of craniosynostosis (up to 75%) and tends to have multi-suture involvement.70,71
Crouzon syndrome is caused by gain-of-function variants in the FGFR2 gene and thus this condition also has impaired and reduced TNALP production. 72 A mouse model of Crouzon syndrome demonstrated a reduction in the craniosynostosis phenotype by increasing serum ALP through TNALP gene expression. 73 Calcification of fibrous tendons (enthesopathies) is a common complication in XLH. Tendons express high levels of FGFR3, which mediates chondrocyte proliferation and differentiation, and aberrant FGFR3 signaling may contribute to inappropriate calcification. 74 Inappropriate FGFR3-mediated signaling in the skull could explain some of the observed phenotypes. The role of TNALP in bone mineralization and soft tissue calcification may help explain the phenotype of XLH, with impaired bone mineralization and calcification of the fibrous layers between sutures. It may also explain premature craniosynostosis with a local increase in PPi and abnormal calcification of the cartilage in the suture space. Further studies should aim to elucidate the PHEX regulation of TNALP and TNALP’s role in aberrant calcification and craniosynostosis in XLH.
Heparan sulfate and local FGF23 expression
Heparan sulfate helps stabilize FGF23–FGFR1 interactions and could contribute to stability and signaling of FGF23–FGFR1.75,76 Loss of heparan sulfate function causes dysfunctional chondrocytes with bony tumors (osteochondromas), as seen in Multiple Hereditary Exostoses (OMIM 13370 and 133701). Autopsies of rachitic skulls have shown osteophyte growth and irregular bone surfaces at the suture line. 77 This is consistent with the inappropriate distribution of FGF23, due to the lack of retention by heparan sulfate causing ectopic signaling, leading to bony growth at the skull’s growth plates. These skull osteophytes form along disorganized rachitic growth plates, such as the osteochondroma growth seen in Multiple Hereditary Exostoses. Osteophytes could potentially bridge the narrow gap between suture spaces, contributing to premature fusion. Investigation of heparan sulfate signaling in craniosynostosis of Hyp mice or patients with XLH could confirm this.
Craniosynostosis in XLH – conclusions and future studies
The cellular mechanisms mediating craniosynostosis in XLH remain unknown. FGFR may both be an upstream and downstream regulator of FGF23 rather than being only the downstream regulator of increased FGF23 (as traditionally assumed). Loss of TNALP activity or expression may lead to increased PPi and altered mineralization, causing craniosynostosis. Heparan sulfate loss of function can cause growth plate osteochondromas and impair the FGF23–FGFR1 interaction. This suggests a requirement for precise local regulation of signaling.
Many questions remain about the cellular basis of craniosynostosis in XLH. Is craniosynostosis a result of the fundamental mineralization defect in XLH, resulting in disorganized suture formation and inappropriate mineralization? Or perhaps craniosynostosis is caused by altered mechanotransduction due to osteomalacia, with compensatory calcification fusion in an effort to overcome the lateral expansion of the skull plates, much like enthesopathies accommodate for tendon insertion in osteomalacic bone. 78 What is the role of dysfunctional paracrine/autocrine signaling of FGFRs and ASARMs to cause craniosynostosis (and further, is craniosynostosis more on the spectrum of a skeletal dysplasia rather than a mineralization defect)? It is unclear why the sagittal suture is predominantly affected, yet some individuals have multiple or even pan-suture involvement. Furthermore, the rates of craniosynostosis in XLH have been reported to be quite high (up to 59%) yet many children do not require neurosurgical intervention; what contributes to the severe phenotype associated with neurological sequelae in some children, but not in others? How does the clinician predict which patients to monitor more closely? It remains to be seen if Burosumab, an anti-FGF23 antibody and novel treatment for XLH, prevents craniosynostosis and further, whether prevention efforts must start early to be maximally (or even minimally) effective. Finally, to date, the prevalence of craniosynostosis has been limited to retrospective studies at tertiary centers;12,13,47 whether these prevalences are representative of other centers with potentially different screening and medical treatment (of hypophosphatemia) practices remain unknown. These unanswered questions reflect the necessity for ongoing research in this area to better understand the cellular mechanisms implicated in XLH and their clinical sequelae, and to guide best practices.
Summary
XLH is due to pathogenic variants causing a loss of function in PHEX, altering FGF23 expression resulting in inappropriately normal or elevated serum FGF23 levels. At the same time, ASARMs are increased, which act as potent focal inhibitors of mineralization. Thus, while FGF23 is the key hormone causing the biochemical abnormalities in XLH including hypophosphatemia, renal phosphate-wasting, and low/normal 1,25 dihydroxyvitamin levels, elevated FGF23 in sutures might signal directly through FGFR1–3 leading to altered bone formation/remodeling directly. The current working models are likely gross simplifications of intricate cellular dysfunctions that occur in XLH. Craniosynostosis is not only an under-recognized complication of XLH with significant clinical sequelae, understanding the cellular mechanisms leading to craniosynostosis in XLH may provide novel options for the management and prevention of craniosynostosis.
Footnotes
Acknowledgements
Dr LMW is a Tier 1 Canada Research Chair in Pediatric Bone Disorders. RTA is a Stollery Science Laboratory Distinguished Researcher and the Canada Research Chair in Renal Epithelial Transport Physiology.
Authors’ Contributions
All authors participated in the review of the article. CG and RTA wrote the article. LMW and DG reviewed the article and contributed significant expert review.
Declaration Of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Dr CG’s research program is supported by the Women and Children’s Health Research Institute and the University of Alberta. Research in the RTA laboratory is funded by the grants from the Women and Children’s Health Research Institute, which is supported by the Stollery Children’s Hospital Foundation, the Canadian Institutes of Health Research, the Kidney Foundation of Canada, and the National Sciences and Engineering Research Council of Canada.