
Abstract
Immune-mediated epilepsies represent an etiologically unique category defined by immune activation within the central nervous system that initiates a pathophysiological cascade resulting in seizures, encephalopathy, and sometimes epileptogenesis with long-term epilepsy. Once considered rare and largely confined to paraneoplastic or postinfectious syndromes, immune-mediated and inflammatory mechanisms are now recognized across a broad spectrum ranging from acute autoimmune encephalitis with reversible seizures, new-onset refractory status epilepticus, or febrile illness-related refractory status epilepticus, to chronic autoimmune-associated epilepsy and drug-refractory structural epilepsies. This review integrates historical perspectives, mechanistic insights from animal and human studies, and contemporary clinical frameworks. It synthesizes advances in neuroimmunology, clinical phenotyping, diagnostics, immunomodulatory and antiseizure therapies, neuromodulation, and patient and family centered outcomes, and outlines future directions focused on biomarker-driven precision medicine, disease-modifying strategies, and interdisciplinary care models.
Introduction
Epilepsy has long been observed in association with inflammatory and immune disorders of the central nervous system (CNS). In the International League Against Epilepsy framework, an immune etiology is invoked when seizures arise as a core manifestation of an immune disorder. 1 The earliest human example is Rasmussen's encephalitis, first described in 1958, where progressive unilateral epilepsy was linked to chronic focal inflammation, reactive gliosis, and neuronal loss. 2 The recognition of paraneoplastic limbic encephalitis and the identification of intracellular neuronal antibodies (eg, anti-Hu and anti-Ma2) reinforced the role of cytotoxic T-cell-mediated immune injury in chronic epilepsy and neurological decline,3,4 while the discovery of neuronal surface antibody-mediated encephalitides, beginning with anti-N-methyl-D-aspartate receptor (anti-NMDAR) encephalitis, demonstrated that seizures can arise from functional immune disruption of synaptic signaling and can be reversible with timely immunotherapy.5,6 In parallel, experimental studies established that acute brain injuries and seizures themselves activate innate immune pathways in the brain—including interleukin-1 receptor 1 (IL-1R1) and toll-like receptor (TLR) signaling, release of danger signals such as HMGB1 and ATP, and activate downstream neuroinflammatory cascades (eg, cytokines, chemokines, prostanoids, and complement factors)—creating a bidirectional “seizure-inflammation” loop.7,8
These advances support a model in which (i) acute symptomatic seizures occur during active CNS inflammation (often antibody-mediated and immunotherapy responsive), while (ii) autoimmune-associated epilepsy (AAE) reflects a persistent seizure predisposition after immune injury or in the setting of enduring immune dysregulation, frequently evolving into drug-resistant network epilepsy.
Neuroinflammation in Structural Epilepsies
In drug-resistant focal epilepsies, histopathological and molecular analyses of surgically resected epileptic foci demonstrate induction of cytokines, chemokines, and downstream inflammatory mediators9–11 in reactive microglia and astrocytes, as well as in neurons and cell components of the blood–brain barrier (BBB). Evidence for inflammation in genetic epilepsies (eg, progressive myoclonus epilepsies and absence epilepsy) and developmental and epileptic encephalopathies is supported by inflammatory signatures in serum and cerebrospinal fluid (CSF) in humans and corresponding animal models, and by seizure responsiveness to therapies with anti-inflammatory activity.12–15
Neuroinflammation in structural epilepsy is primarily driven by innate immunity activation within brain parenchymal cells and is associated with extravasation of macrophages and neutrophils. The presence of adaptive immunity cells (T and B cells) depends on the epilepsy etiology.7–12 In particular, a convergent triad of ictogenic inflammatory signaling involves: (i)
Key Cellular Mechanisms Underlying the Effects of Neuroinflammation on Epilepsy Outcomes.

Abbreviations: NMDA, N-methyl-D-aspartate; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; BBB, blood–brain barrier; IgG: immunoglobulin.
Transcriptomic profiling has revealed activation of an immunotranscriptome—an inflammatory gene program—across pediatric and adult focal epilepsies.18,19 In temporal lobe epilepsy (TLE), neuroinflammation is present even in nonsclerotic hippocampi (eg, lesion-associated TLE), involving predominantly reactive astrocytic programs and correlating with seizure activity; however, seizures are not the sole determinant. Tissue histopathology also shapes inflammatory burden: inflammation is typically higher in hippocampi with sclerosis than in nonsclerotic hippocampi, and greater in focal cortical dysplasia (FCD) IIb than FCD I, even in patients with similar seizure burden.9,10,20
Neuroinflammation is also bidirectionally linked with mammalian Target of Rapamycin pathway activation in structural epilepsies 16 due to an inefficient resolution mechanism: anti-inflammatory mediators (eg, IL-1Ra, IL-10, pro-resolving lipid mediators, and CD47/CD200 signaling) may be either insufficient relative to pro-inflammatory activation or intrinsically ineffective. Dysregulation of transcription factors and epigenetic mechanisms (promoter methylation and microRNAs) further biases toward persistent inflammatory gene activation.8,21,22
Biomarkers of neuroinflammation include positron emission tomography (PET) imaging of translocator protein ligands for reactive glia, monoamine oxidase B-targeted tracers (eg, deprenyl) in reactive astrocytes, and magnetic resonance spectroscopy measures (eg, myoinositol). 23
NORSE/Febrile Infection-Related Epilepsy Syndrome (FIRES)
NORSE is defined as new-onset refractory status epilepticus in a patient without prior epilepsy or relevant neurological disorder, without a clear acute structural, toxic, or metabolic cause; FIRES requires a febrile infection 2 weeks to 24 h before onset. 24 The reported incidence estimates are ∼0.65/100,000 adults and ∼1/million children.25,26 Inflammation contributes to antiseizure medication (ASM) resistance, and blockade of IL-1 or IL-6 signaling has shown translational promise in NORSE/FIRES 27 consistent with observations of cytokine storm signatures and limbic neuroinflammation in these syndromes. 28 NORSE and FIRES may result in AAE as the chronic consequence.
Epidemiology and Disease Concepts in Autoimmune-Mediated Epilepsies
An immune etiology is conceptualized as autoimmune-mediated CNS inflammation; however, in autoimmune encephalitis, seizures may resolve with treatment and not necessarily confer a long-term epilepsy predisposition. 29 Determining when persistent seizures transition from acute symptomatic seizures to epilepsy remains a key challenge for incidence estimation.
Acute symptomatic seizures secondary to autoimmune encephalitis are commonly associated with neuronal surface antibodies (eg, NMDAR and leucine-rich glioma-inactivated 1 [LGI1]). They are typically immunotherapy responsive and often resolve with adequate treatment30,31; although some cell surface antibodies (LGI1, gamma-aminobutyric acid type A receptor [GABAAR], and gamma-aminobutyric acid type B receptor [GABABR]) are associated with an increased risk of subsequent epilepsy (Table 1). Rada and Bien 32 proposed a practical definition of autoimmune encephalitis-associated epilepsy requiring: (1) prior diagnosis of autoimmune encephalitis due to cell-surface antibodies, (2) persisting seizures ≥2 years after immunotherapy, (3) no encephalitis activity on magnetic resonance imaging (MRI) and no fluorodeoxyglucose-PET (FDG-PET) hypermetabolism, (4) normal CSF cell count, and (5) substantial antibody titer reduction. In contrast, AAE is typically observed in cytotoxic T-cell-mediated Rasmussen syndrome or in epilepsies linked to intracellular antigens, resulting in durable structural or network changes and resistance to immunotherapy. 30
The AAE frequency estimates vary depending on cohorts and testing practices. In epilepsy of unknown etiology, serum antibodies suggestive of autoimmune contribution have been reported in 3.4% to 10.5% of patients,33,34 though CSF confirmation and clinical correlation are inconsistent, and misdiagnosis (such as antibodies with limited clinical specificity) remains a concern.35,36 Recent population-based data also show that autoimmune seizures and epilepsies are rare, with a prevalence of about 10 per 100,000, and AAE prevalence of approximately 7 per 100,000; however, they contribute disproportionately to the overall disability burden at the population level, as reflected by disability-adjusted life years. 37
Clinical Manifestations and Diagnosis of Immune-Mediated Epilepsy
Seizures are one of the cardinal features of immune-mediated CNS disease, including autoimmune encephalitis, and can be the predominant presentation in some patients (Table 2). Clinically, immune-mediated seizures are often discussed in 2 temporal and mechanistically distinct categories: (1) Acute symptomatic seizures secondary to autoimmune encephalitis, an acute manifestation of immune dysregulation often with good response to immune therapy, and (2) a chronic, typically medically refractory AAE.30,32 AAE can be further stratified according to antibody-associated phenotypes (Table 3). The following are the three most common antibodies associated with immune-mediated epilepsy and seizures.
When to Suspect Immune-Mediated Seizures/Epilepsy.

Abbreviations: FAS, focal aware seizure; FIAS, focal impaired awareness seizure; MTS, mesial temporal sclerosis; EEG, electroencephalogram; MRI, magnetic resonance imaging; FLAIR, fluid-attenuated inversion recovery.
Clinical Characteristics of Most Common Antibody-Associated Epilepsies.
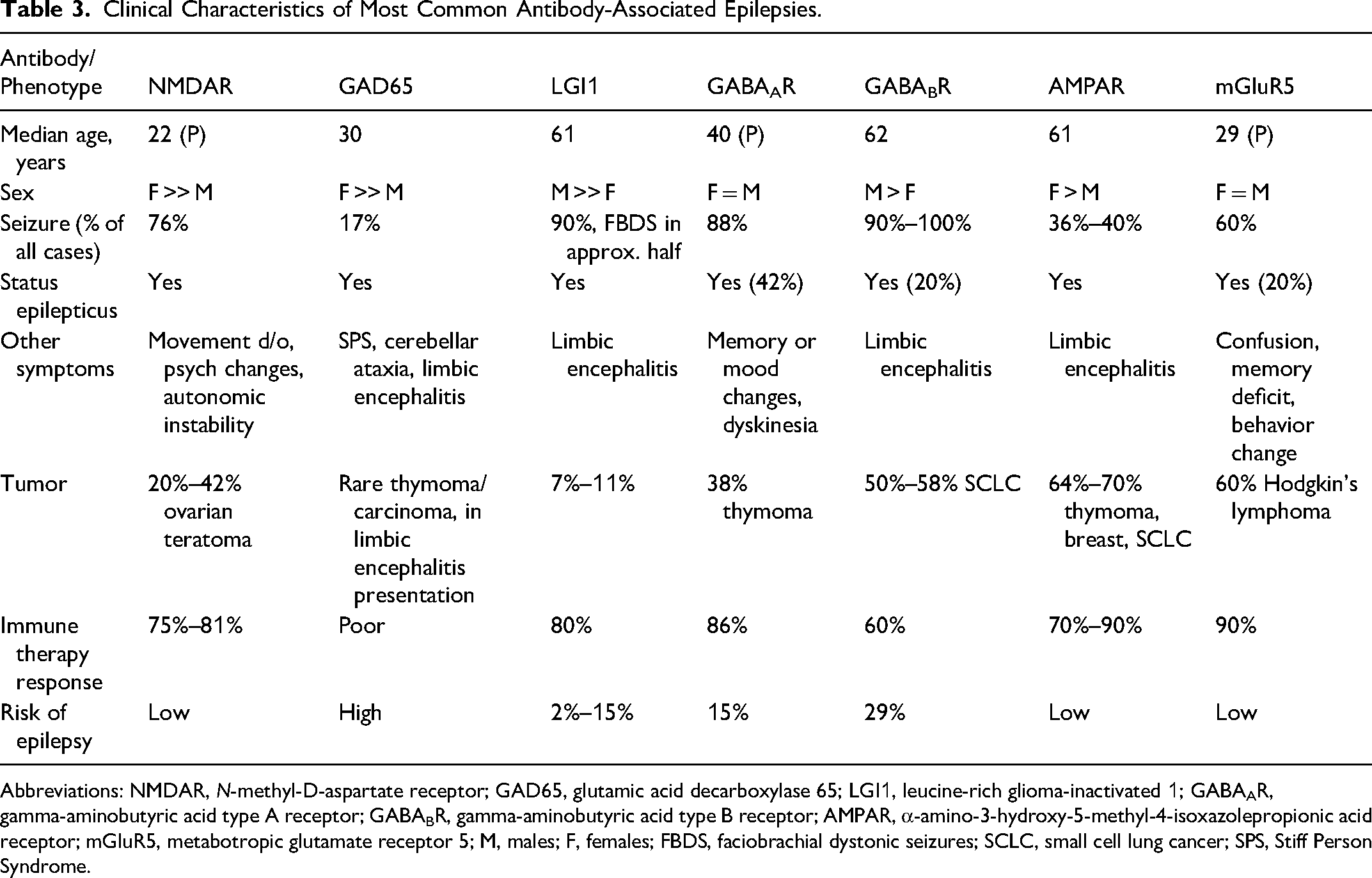
Abbreviations: NMDAR, N-methyl-D-aspartate receptor; GAD65, glutamic acid decarboxylase 65; LGI1, leucine-rich glioma-inactivated 1; GABAAR, gamma-aminobutyric acid type A receptor; GABABR, gamma-aminobutyric acid type B receptor; AMPAR, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; mGluR5, metabotropic glutamate receptor 5; M, males; F, females; FBDS, faciobrachial dystonic seizures; SCLC, small cell lung cancer; SPS, Stiff Person Syndrome.
Anti-NMDAR encephalitis often affects young adults with female predominance and may cause psychiatric and cognitive features, seizures, movement disorders, autonomic instability, and reduced consciousness.5,6 Triggers may include ovarian teratoma or post-herpes simplex encephalitis (6). Seizures can occur early and may progress to status epilepticus; long-term epilepsy risk is relatively low, with high rates of seizure freedom after effective immunotherapy (6).
LGI1 encephalitis commonly manifests with faciobrachial dystonic seizures (FBDSs) and seizures with autonomic features (piloerection and ictal bradycardia), psychic and sensory symptoms or dizziness, cognitive changes, and hyponatremia. Prompt immunotherapy reduces seizure burden and may mitigate cognitive impairment. While patients typically respond to immunotherapy, the risk of developing AAE ranges from 2% to 15%.38,39
Glutamic acid decarboxylase 65 (GAD65)-associated epilepsy median age of onset in the third decade of life, with a marked female predominance (∼84%) 40 commonly co-occurs with other systemic autoimmune disorders.41–43 The typical clinical course is characterized by chronic, drug-resistant epilepsy, most often with temporal and perisylvian seizure onset zones. Distinctive clinical features include a high seizure burden, temporal lobe seizure semiology, autonomic manifestations such as piloerection, experiential symptoms including déjà vu and anxiety, and reflex phenomena such as musical auras and musicogenic seizures. 44 High-titer GAD65 antibodies defined as >10,000 IU/mL or >20 nmol/L may account for up to 10% of adult-onset TLE of unknown etiology 45 (Figure 1).
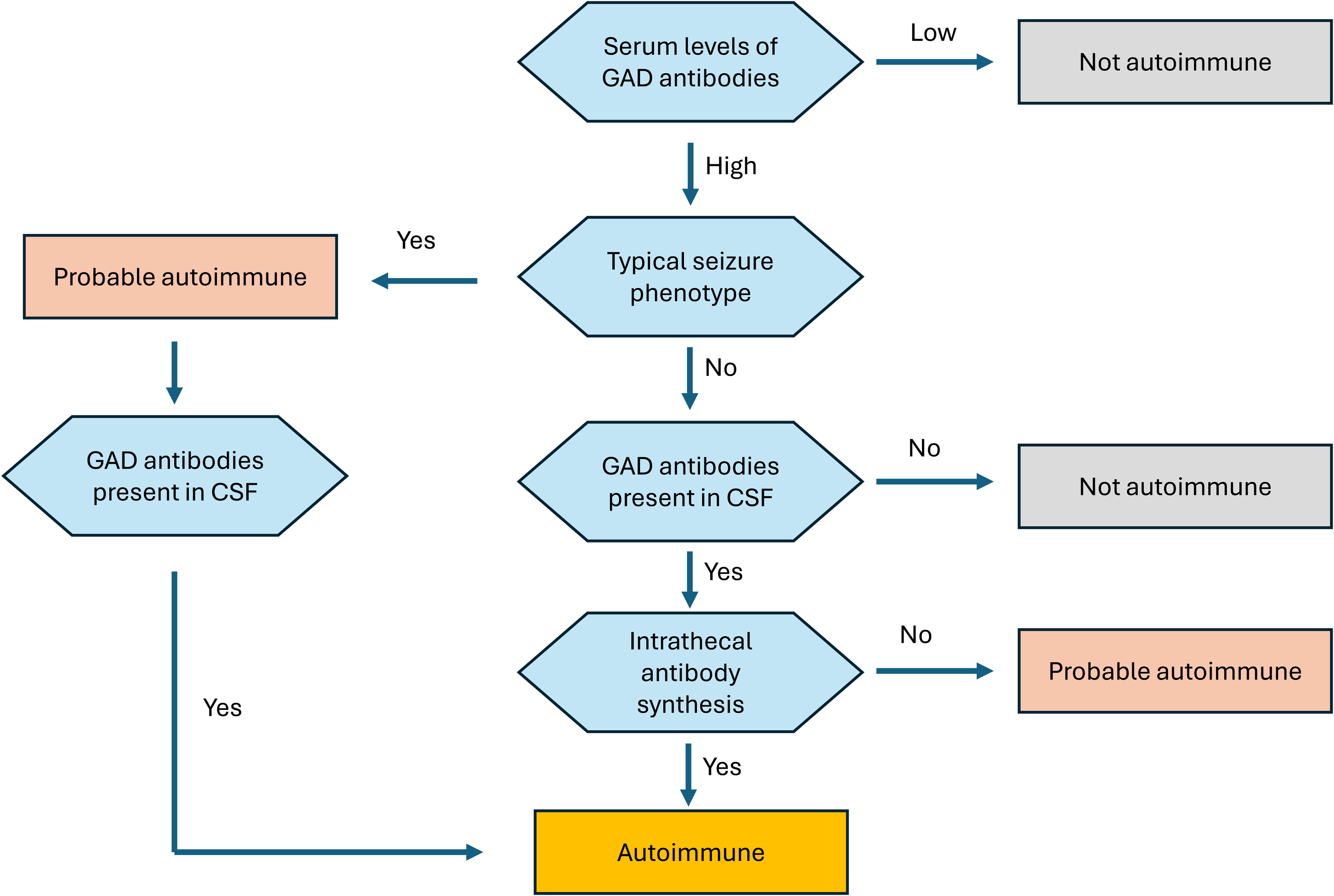
Diagnostic decision tree in nonlesional medically refractory temporal lobe epilepsy with seizure phenotype suggestive of possible glutamic acid decarboxylase 65 (GAD65) antibody-mediated etiology.
In contrast, low-titer GAD65 antibodies (<2000 IU/mL or <2 nmol/L) are commonly detected in individuals with systemic autoimmune diseases and may also be found in healthy individuals. Therefore, careful interpretation of antibody titers is essential, and in selected cases, paired serum and CSF testing may help clarify clinical relevance.
Electroencephalogram (EEG) findings in AAE are variable but may reveal high subclinical seizure burden, bilateral independent temporal discharges, and multifocal onset zones; in some adult patients, hyperventilation may provoke focal seizures. 30 Neuroimaging may be normal early or show medial temporal T2/fluid-attenuated inversion recovery (FLAIR) hyperintensity in limbic encephalitis, progressive hemispheric atrophy in Rasmussen syndrome, multifocal cortical-subcortical changes in gamma-aminobutyric acid type A (GABAA), myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD), or anti-Hu, and basal ganglia involvement in LGI1. FDG-PET can improve early sensitivity and may show characteristic regional patterns (3).
Predictive models and scores have been developed to assist in diagnosing AAE and identifying patients who would benefit from antibody testing, including: the antibody prevalence in epilepsy and encephalopathy score,35,46 the Oxford group score, 34 and the antibodies contributing to focal epilepsy signs and symptoms score. 33
Neuronal antibody testing is essential for confirmation and phenotypic stratification but requires careful interpretation. Broad panels are generally preferred due to phenotypic overlap. When feasible, testing should precede immunotherapy: plasma exchange (PLEX) can cause false negatives, and intravenous immunoglobulin (IVIG) can yield false positives. Both serum and CSF should be tested when possible—CSF is more sensitive/specific for most neuronal surface antibodies, while serum is often more sensitive for LGI1/CASPR2 and glial antibodies. A subset of patients will have seronegative autoimmune encephalitis. These patients are more likely to present with status epilepticus, and a subset will develop AAE. Careful application of the consensus criteria will help in the identification of these patients. 47
Pitfalls include isolated serum positivity without compatible clinical features or CSF support. Several antibody findings are frequently nonpathogenic in seizure populations.
Medical Treatment: Targeting the Immune System in Acute Symptomatic Seizures and Immune-Mediated Epilepsy
Early immunotherapy is associated with improved seizure control and cognitive outcomes in acute symptomatic seizures.6,35 While a minority of patients with antibody positivity but limited encephalitic features may have transient improvement with ASMs alone, most benefit from immunomodulation. Evidence for immunotherapy efficacy in chronic AAE is more limited and largely derived from retrospective cohorts and case series, with few randomized trials.
For patients with LGI1 or CASPR2 antibody-associated encephalitis, the current consensus recommends initial treatment with high-dose intravenous methylprednisolone (IVMP), with IVIG or PLEX considered in specific scenarios, such as corticosteroid intolerance or contraindication.48,49
In NMDAR antibody-associated encephalitis, the recommended acute-phase regimen involves IVMP in combination with IVIG or PLEX, reflecting the need for more aggressive immunomodulation. 50 For GAD65 antibody-associated autoimmune epilepsy, either IVMP or IVIG may be used as first-line therapy, especially within 2 years of seizure onset or in the limbic encephalitis phenotype.44,50 Patients with GABABR or GABAAR antibody-associated encephalitis generally receive IVMP combined with IVIG or PLEX.51,52 In cases of high-risk paraneoplastic antibody-associated encephalitis, IVMP remains the cornerstone of acute-phase treatment. The 12-week protocol consists of 1 g IV daily for three consecutive days, followed by weekly infusions for 5 weeks, then every 2 weeks for 6 weeks; the 6-week protocol consists of 1 g IV daily for 3 days, followed by weekly infusions for 5 weeks.50,53 For some patients, an IVIG treatment trial could also be considered. IVIG protocols mirror those of IVMP, with dosing at 0.4 g/kg daily for 3 days, followed by weekly or biweekly 0.4 g/kg infusion.41,50
Second-line therapies include rituximab, cyclophosphamide, mycophenolate mofetil, azathioprine, tocilizumab, and bortezomib.41,50 Rituximab is often utilized during the acute phase of NMDAR immunoglobulin G (IgG) autoimmune encephalitis. Evidence from cohort studies indicates that rituximab (375 mg weekly × 4 or 1 g twice, 2 weeks apart) significantly improved functional outcomes and reduced relapse rates.6,54 Combination therapy (rituximab + cyclophosphamide) may be considered in severe or refractory cases (6). Similarly, in LGI1 IgG autoimmune encephalitis, chronic immunotherapy with agents such as rituximab significantly reduces relapse rates. 54
Tocilizumab, an IL-6 receptor antagonist, has emerged as a promising option for refractory autoimmune encephalitis 55 to achieve meaningful clinical improvement in autoimmune encephalitis patients unresponsive to first-line and B-cell depleting therapies. In these cohorts, approximately 60%–70% of patients demonstrated functional recovery and seizure reduction. The study emphasized early escalation to tocilizumab, ideally within 1 month of rituximab failure, to optimize outcomes and reduce long-term disability. A teratoma removal + steroids + IVIG + rituximab + tocilizumab (T-SIRT) protocol has also been proposed for severe NMDAR IgG autoimmune encephalitis. 56
Neonatal Fc receptor (FcRn) blockade is another emerging immunotherapy to be evaluated in AAE. In controlled studies for other autoimmune neurological indications, FcRn inhibitors such as efgartigimod significantly reduced pathogenic IgG levels and improved clinical outcomes. 57
Cyclophosphamide has a specific role in high-risk paraneoplastic or intracellular antibody-associated autoimmune seizures with an aggressive disease course and poor response to first-line immunotherapy. In such cases, early introduction of cyclophosphamide has been associated with improved seizure control and stabilization of neurological decline. 17 Since many of these patients have an underlying malignancy, treatment planning often requires close coordination with the oncology team. Chemotherapy for cancer, when combined with cyclophosphamide, can lead to bone marrow suppression. 58
Monitoring treatment response in autoimmune seizures requires both short-term and long-term strategies. In the first 3–6 months, clinicians should track seizure frequency, perform regular neurological and mental status examinations, and, in some scenarios, schedule an ambulatory EEG to detect subclinical seizures. 46 Imaging with brain MRI and, when indicated, FDG-PET should be repeated to assess residual inflammation.50,53 Outcome scales such as the modified Rankin Scale for global disability, the Clinical Assessment Scale in Autoimmune Encephalitis, or the anti-LGI1 autoimmune encephalitis outcome rating score or LGI1-Antibody Encephalitis Rating scale for disease severity, can be used early to quantify baseline impairment and monitor progress.59–61 Long-term monitoring focuses on relapse prevention and cognitive outcomes through neuropsychological evaluations, neuropsychiatric assessments, and standardized disease severity scales.
Surgical and Neuromodulatory Treatment Strategies in AAE
AAE reflects both immune-mediated injury and secondary structural reorganization. 62 Surgical evaluation is indicated when patients remain refractory to immunotherapy and optimized antiseizure medications or when a persistent focal epileptogenic lesion is evident following the resolution of acute inflammation. 62 In cases with clearly localized pathology or frequent, disabling seizures, earlier evaluation may be justified. Even when complete seizure freedom is unlikely, surgery may provide meaningful palliative benefit and, in some cases—such as glutamic acid decarboxylase-associated TLE (GAD-TLE)—may represent the only curative option. 40 Presurgical evaluation follows standard protocols for focal medically refractory epilepsy, but often requires intracranial EEG due to multifocality and the possibility of microscopic or radiographically occult lesions. 63 Here we focus on surgical and neuromodulatory approaches for AAE secondary to autoimmune or infectious encephalitis, while other immune-mediated epilepsies, such as Rasmussen syndrome, are not addressed.
Table 4 summarizes clinical outcomes in AAE cohorts treated with surgical and neuromodulatory interventions.40,64–75 The data are limited as there have been no randomized controlled trials comparing surgery with continued medical therapy in AAE. Existing data are confounded by heterogeneity in antibody profiles, timing relative to inflammation, and variable follow-up. 40 Most surgical experience comes from GAD-TLE cohorts, where temporal lobectomy achieves seizure freedom in only 15%–25% of patients.40,64,65 Lower success rates compared with other lesional epilepsies likely reflect persistent inflammation, broader epileptogenic networks such as temporal-plus epilepsy, or bilateral temporal involvement.
Clinical outcomes from representative studies evaluating surgical and neuromodulatory interventions in patients with autoimmune-associated epilepsy.
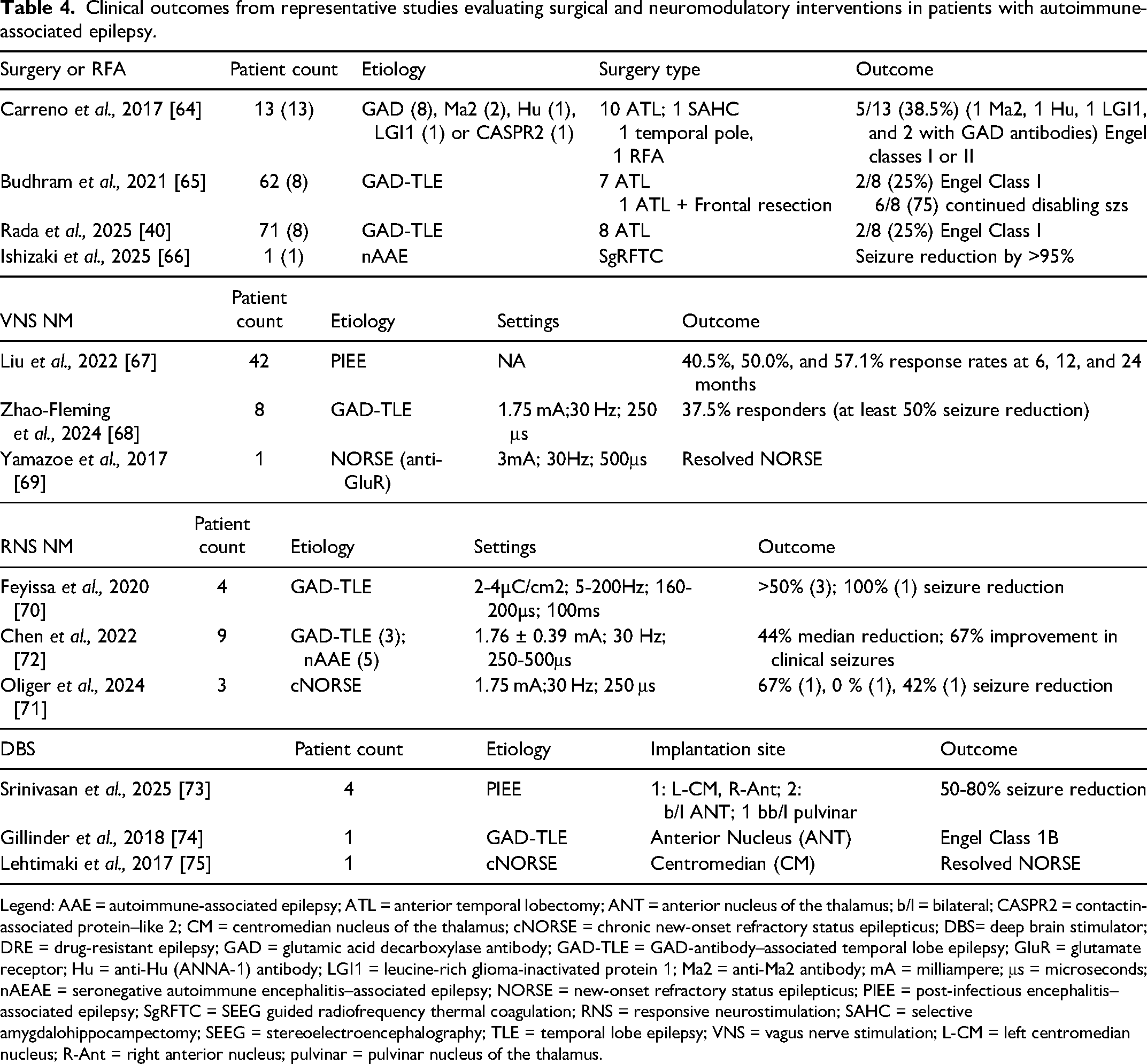
Legend: AAE = autoimmune-associated epilepsy; ATL = anterior temporal lobectomy; ANT = anterior nucleus of the thalamus; b/l = bilateral; CASPR2 = contactin-associated protein–like 2; CM = centromedian nucleus of the thalamus; cNORSE = chronic new-onset refractory status epilepticus; DBS= deep brain stimulator; DRE = drug-resistant epilepsy; GAD = glutamic acid decarboxylase antibody; GAD-TLE = GAD-antibody–associated temporal lobe epilepsy; GluR = glutamate receptor; Hu = anti-Hu (ANNA-1) antibody; LGI1 = leucine-rich glioma-inactivated protein 1; Ma2 = anti-Ma2 antibody; mA = milliampere; μs = microseconds; nAEAE = seronegative autoimmune encephalitis–associated epilepsy; NORSE = new-onset refractory status epilepticus; PIEE = post-infectious encephalitis–associated epilepsy; SgRFTC = SEEG guided radiofrequency thermal coagulation; RNS = responsive neurostimulation; SAHC = selective amygdalohippocampectomy; SEEG = stereoelectroencephalography; TLE = temporal lobe epilepsy; VNS = vagus nerve stimulation; L-CM = left centromedian nucleus; R-Ant = right anterior nucleus; pulvinar = pulvinar nucleus of the thalamus.
Stereoelectroencephalography-guided radiofrequency thermocoagulation (SgRFTC) has emerged as a promising alternative in a case affected by AAE that demonstrated >95% seizure reduction after SgRFTC. 66 By targeting deep or distributed networks and creating multiple strategic lesions, SgRFTC broadens surgical options for AAE patients ineligible for resection due to a multifocal epileptogenic process. Early outcomes are promising, but multicenter studies are urgently needed to define the role of this intervention.
Neuromodulation includes vagus nerve stimulation (VNS), responsive neurostimulation (RNS), and deep brain stimulation (DBS), and is a promising alternative for AAE when resection or ablation is not feasible due to diffuse or poorly localized epileptogenic networks. The mechanisms underlying its therapeutic effects in AAE remain incompletely understood but may involve modulation of inflammation, glial signaling, pathological network dynamics, and the effect of improved seizure control.76,77 However, heterogeneity in patient selection, implantation timing, neuromodulatory approaches, and others limits conclusions.70,72–74,76 Interestingly, immunotherapy does not seem to increase the risk of device-related infection.70,72–74,76 RNS provides both therapeutic benefit and long-term intracranial EEG monitoring, which can be especially useful in cases with multifocal seizures (Figure 2) and in informing immunotherapy use and medication adjustments. The RNS data can also help distinguish nonepileptic events in patients affected by high seizure burden and cognitive or psychiatric comorbidities. Evidence for DBS application remains limited.70,72–74,76
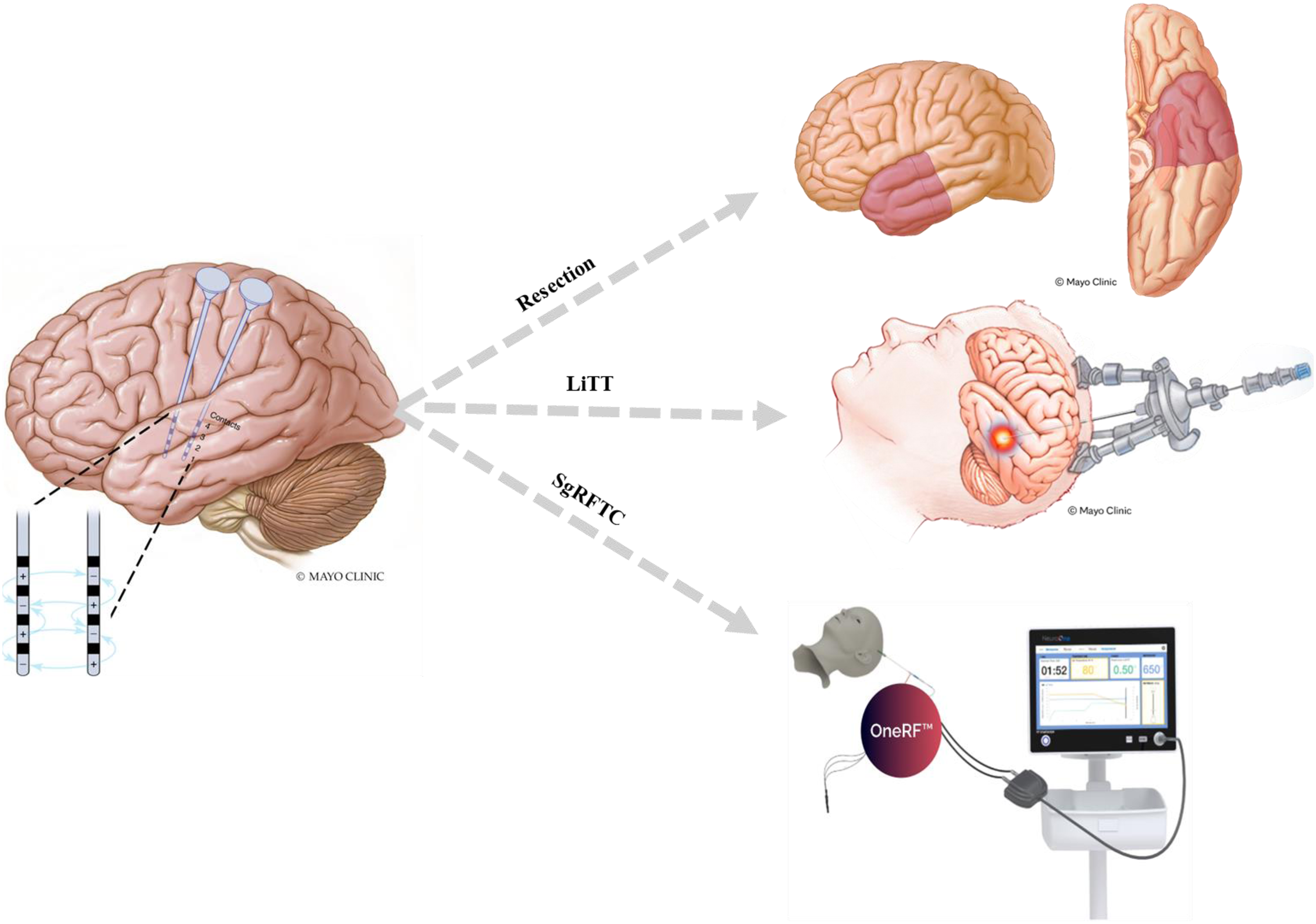
Staged approach using chronic electrocorticography with the RNS system to localize the predominant seizure focus, guiding selection of resective or ablative therapies—including LiTT or SgRFTC—in patients with autoimmune-associated epilepsy.
Cognitive, Psychiatric, and Patient-Centered Outcomes
Long-term outcomes often extend beyond seizure control. Cognitive and functional outcomes vary by etiology, with consistently worse outcomes in GAD-associated disease and in many NORSE/FIRES survivors. 78 While many patients with antibody-mediated encephalitis achieve seizure freedom, cognitive sequelae and neuropsychiatric symptoms (anxiety, depression, aggression, emotional lability, and apathy) are frequent and reported in ∼30% of adult and up to ∼80% of pediatric survivors. These contribute substantially to caregiver burden and quality-of-life impairment79,80 and underscore that outcomes are shaped not only by biology, but also by communication, systems of care, and research infrastructure. Advocacy-driven efforts have advanced consensus definitions for NORSE/FIRES, treatment recommendations, standardized biospecimen collection, and international case conferencing, helping clinicians communicate prognosis with greater clarity and supporting collaborative research.24,81,82
Summary and Future Directions
Immune-mediated epilepsy exemplifies how immune activation and deregulation can initiate, amplify, and perpetuate epileptic networks. Across the spectrum—from immune-mediated acute symptomatic seizures to chronic AAE—key lessons include: (1) early recognition and treatment during active inflammation improves outcomes; (2) mechanistic distinctions between antibody-mediated and T-cell-driven disease shape prognosis and therapeutic responsiveness; (3) refractory disease frequently reflects network epilepsy requiring consideration for surgical intervention, neuromodulation, and multidisciplinary care; and (4) AAE is a chronic and multifaceted condition with often complex neuropsychiatric comorbidities with long-term impact on patients and their families thus requiring ongoing communication and multidisciplinary care. Future progress will depend on validated biomarkers that distinguish active inflammation from fixed network epilepsy, precision immunotherapy and immune-targeted escalation strategies, and disease-modifying interventions with pathway-specific anti-inflammatory drugs that interrupt seizure—inflammation feedback loops. Equally important is integration of cognitive, psychiatric, and family centered outcomes into trials and care pathways, recognizing immune-mediated epilepsy as a translational framework with implications across the broader epilepsy spectrum.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.