
Abstract
Ehlers-Danlos syndrome (EDS) represents a group of inheritable connective tissue disorders. Patients with type IV or vascular EDS, autosomal dominant pattern of inheritance, may present with aneurysm formation or arterial dissection. Due to vessel fragility, operative therapy for such disorders has been reserved for compelling indications in which benefit clearly warrants risk, yet assessment of risk is largely clinical with operative decisions guided by factors such as response to previous operations and age at onset of index vascular complications. We present 2 patients with differences in their clinical presentations and outcomes and review the literature with emphasis on operative decision making.
Introduction
Ehlers-Danlos syndrome (EDS) is a group of inherited connective tissue disorders variably affecting skin, joints hollow viscera, and blood vessels caused by a defect in the synthesis of collagen. 1 Vascular or type IV EDS is an autosomal dominant defect in type III collagen synthesis affecting approximately 1 in 100 000 to 250 000 people. 2 Mutations in COL3A1 have been identified. Vascular EDS is considered one of the most serious forms of EDS because blood vessels and organs are prone to tearing (rupture). Due to vessel fragility, elective operations in patients with vascular EDS has been reserved for compelling indications in which benefit clearly warrants risk, yet assessment of risk is largely clinical with operative decisions guided by factors such as response to previous operations and age at onset of index vascular complications.
Case Report 1
A 52-year-old male presented with multiple daily episodes of dizziness and blurring of vision in the left eye. These symptoms had escalated in frequency and severity over a period of 3 months. The patient had documented vascular EDS with mutation of COL3A1 gene. Family history was relevant with mother and younger brother diagnosed with EDS with the brother dying at age 32 from a ruptured thoracic aortic aneurysm. The 74-year-old mother is has not experienced any major medical problems. The patient had experienced a stroke in childhood at age 7 and had required enucleation of his right eye due to trauma when he was 11 years old. He had experienced no excessive bleeding events in his teens or early adulthood. He underwent uneventful right nephrectomy for an 8.5 cm renal adenocarcinoma 2 years prior to his vascular referral. On physical examination, the patient’s skin was mildly but not inordinately thin. Carotid pulses were not palpable and there were no carotid bruits. The brachial artery pulses were normal with equal arm pressures. Carotid duplex ultrasonography (CDU) demonstrated bilateral common carotid artery occlusion with retrograde flow in the left internal carotid artery and antegrade flow in the right internal carotid artery (Figure 1). The arterial flow in both vertebral arteries was antegrade. Computed tomography angiogram (CTA) of the neck and chest confirmed findings of bilateral common carotid artery occlusion with internal carotid patency (Figure 2 ). After reviewing the patient’s past operative history and in consideration of the severity of his symptoms of cerebrovascular insufficiency, we opted to proceed with operative carotid revascularization. This was accomplished with a left subclavian to left internal carotid bypass using a 6 mm heparin-bonded polytetrafluorethylene (PTFE) graft. At surgery, the subclavian and carotid arteries appeared normal and the operation was accomplished with minimal blood loss. The patient recovered uneventfully. Postoperative CDU at 3 and 6 months showed patency of the bypass graft. He has enjoyed complete resolution of symptoms with unilateral bypass, thus the contralateral carotid occlusion has not required intervention.
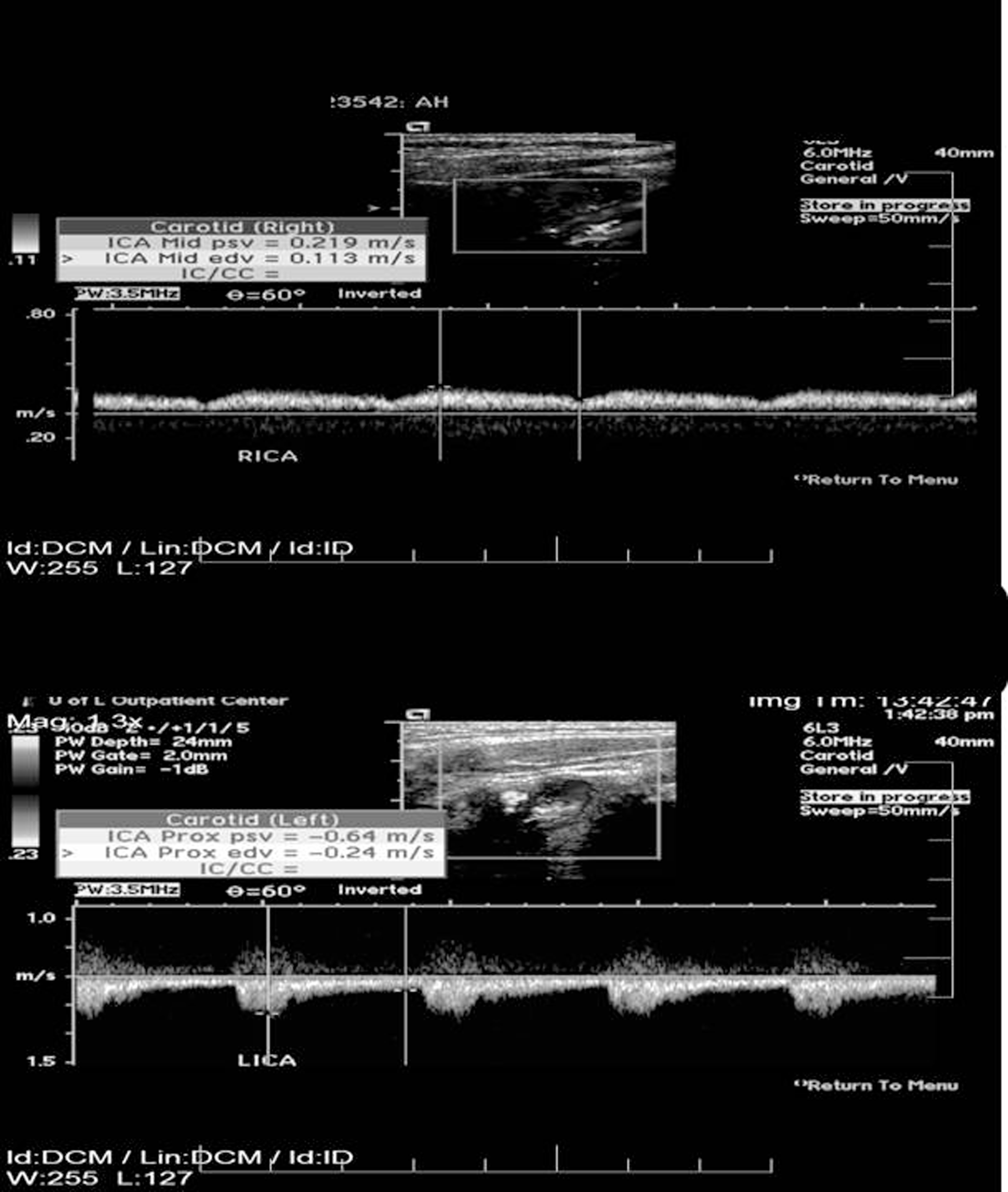
Carotid duplex ultrasonography demonstrates bilateral common carotid artery occlusion with retrograde flow in the left internal carotid artery and antegrade flow in the right internal carotid artery.
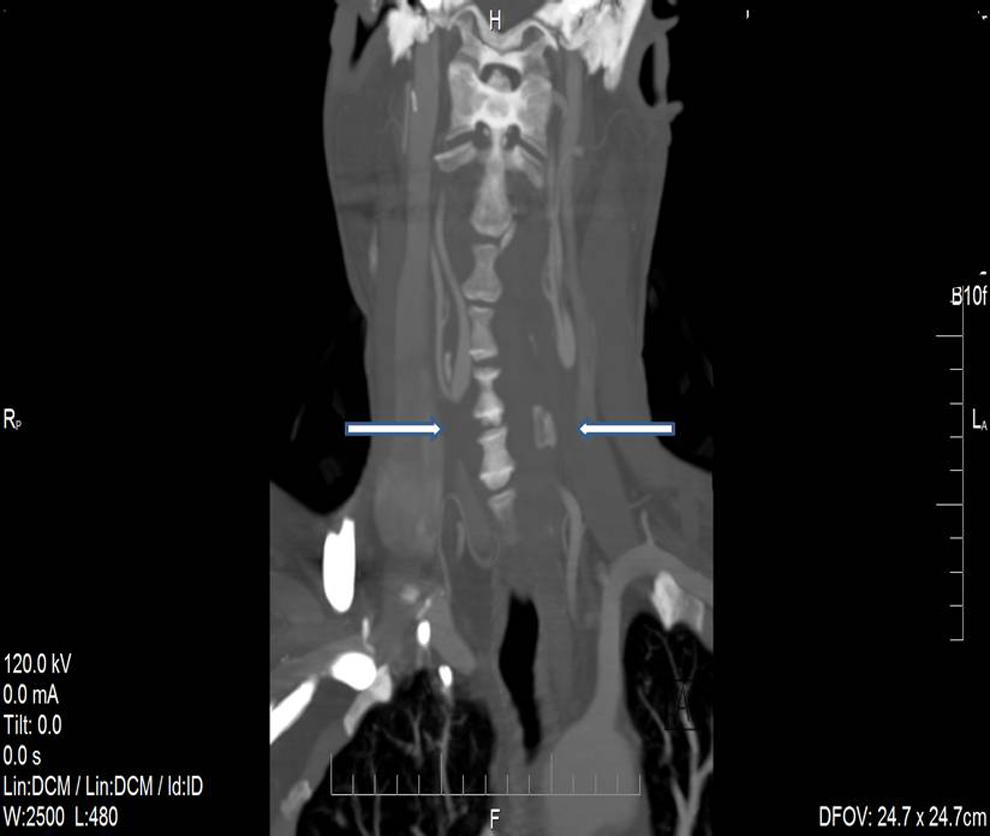
Computed tomography angiogram of the neck and chest confirms findings of bilateral common carotid artery occlusion with internal carotid patency as demonstrated by arrows.
Case Report 2
A 28-year-old male presented with progressive abdominal pain and right flank pain radiating into his right lower extremity. In the emergency department, he became increasingly tachycardiac and hypotensive with right lower abdominal tenderness. Abdominal and pelvic CT scan showed a large right retroperitoneal hematoma with active extravasation from a ruptured right common iliac artery dissection (Figure 3 ). He underwent emergency celiotomy, and common iliac artery was controlled. However, extreme fragility of the artery was recognized and direct revascularization based on aorta was avoided. Instead common iliac artery proximal to the point of rupture and the right proximal external and internal iliac arteries were individually ligated. Review of the CT scan revealed a focal, proximal left internal iliac artery dissection that had not ruptured. We opted not to approach that lesion at the time due to a desire to avoid bilateral simultaneous internal iliac ligation as well as to avoid potential injury to the left common and/or external iliac arteries. Revascularization of right leg was achieved with left-to-right femoral/femoral bypass utilizing 8 mm heparin-bonded PTFE graft.
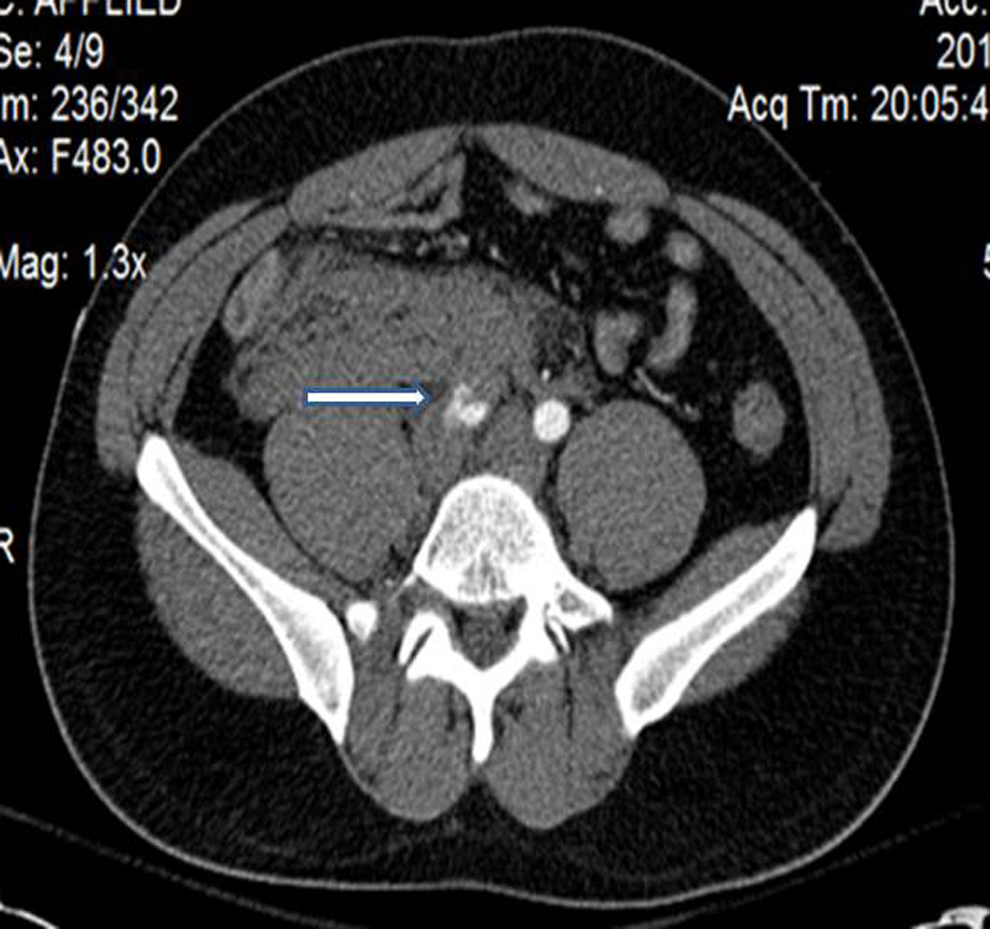
Abdominal and pelvic computed tomographic scan shows a large right retroperitoneal hematoma with active extravasation from a ruptured right common iliac artery dissection as demonstrated by the arrow.
It was later found that the patient had a history of easy bruisability that was never formally investigated. Family history was remarkable for a cousin who died at 32 years of age from an aortic dissection and rupture thought to be related to Marfan syndrome, and another cousin who had been diagnosed with disordered EDS of an unknown type.
Postoperatively patient required tracheostomy due to respiratory failure Three weeks postoperatively, the patient complained of left groin pain. The CTA showed a left retroperitoneal hematoma with stable left iliac artery dissection. The patient became progressively hypotensive and developed cardiac arrest. After resuscitation, angiography was undertaken which failed to demonstrate bleeding from the left internal iliac artery. The patient died the following day and autopsy revealed a stable left iliac artery dissection with spontaneous retroperitoneal hematoma. A left posterior inferior cerebellar artery aneurysm was also identified.
Discussion
Ehlers-Danlos syndrome describes a group of more than 10 different inherited disorders, all of which involve a genetic defect in collagen and connective tissue synthesis and structure.3,4 Ehlers-Danlos syndrome can affect the skin, joints, organs, and blood vessels.5–7 This syndrome is clinically heterogeneous, and the underlying collagen abnormality is different for each type. Clinical recognition of the types of EDS is important.
The term vascular EDS was coined by Barabas.8,9 The clinical diagnosis of vascular EDS is established from at least 2 of 4 major clinical criteria: easy bruising, thin skin with visible veins, characteristic facial features, and rupture of arteries, uterus, or intestines. 10 Although the presence of 2 major clinical criteria is highly suggestive of vascular EDS, laboratory testing for procollagen III deficiency is strongly recommended.11,12 Patients frequently die in their third and fourth decade and survival beyond 50 years is exceptional. 13 Affected patients frequently describe relatives who died suddenly with some vascular etiology. Both our patients had positive family history of members with EDS, although vascular EDS was uncertain.
Vascular EDS usually follows an autosomal dominant pattern of inheritance. It is caused by mutations in the type III collagen gene, COL3A1 leading to diminished levels of type III collagen. Results of biochemical and gene mutation analyses may take months, and operative therapy should not be delayed if indicated. One potential problem is that the nature of the COL3A1 gene mutation does not accurately predict the type, extent, and prognosis of EDS IV. 14 In the analysis of the mutations identified in the COL3A1 gene to this point, at least 2 classes of mutations—substitutions of glycine in the triple helical domain by alanine and introduction of premature termination codons are underrepresented among individuals with vascular EDS. Thus, some mutations in COL3A1 may not produce a vascular EDS phenotype. 15 The patient in case report 1 underwent a successful nephrectomy and subsequently a left subclavian to internal carotid artery bypass without any significant complications. This patient had tested positive for COL3A1 gene. It is likely that this patient did not express the vascular EDS phenotype. The second patient presented with surgical emergency which was managed effectively but still had a dismal outcome consistent with full expression of the vascular EDS phenotype.
The traditional recommendation when dealing with symptoms in a patient with type IV EDS has been a conservative approach, with operative treatment reserved for patients with imminent or frank life-threatening bleeding. 16 However, as depicted in our first case report, the surgeon may be faced with the dilemma of a patient with compelling symptoms that may necessitate operative intervention. It is prudent to consider the patient’s previous surgical history as it may help in decision making. On the other hand, frank bleeding necessitates surgical treatment, yet the incidence of postoperative bleeding complications and late graft-related problems can be significant, including shortened survival in this patient group as was demonstrated by our second patient. Endovascular approaches to the control of bleeding have been reported, but percutaneous and transluminal interventions must be performed cautiously as well.
Although no specific therapies delay the onset of complications in patients with vascular EDS, the role of β-adrenergic blockade for reducing the risk of arterial rupture and complications has been suggested. 17 Genetic counseling with biochemical testing may help in educating the families of patient with type IV EDS.
Conclusion
Vascular EDS is caused by a spontaneous point mutation in the COL3A1 gene that encodes the chains of type III procollagen. Increased tissue and particularly vessel fragility results in a higher risk of intraoperative and late surgical complications; thus, elective surgery for individuals with vascular EDS is generally discouraged. To our knowledge, this is the first reported case of symptomatic bilateral common carotid occlusion in a patient with vascular EDS. Our patient had compelling symptoms which raised concern for imminent stroke, thus prompting a decision to operatively intervene. His surgical course was benign, suggesting that, although positive for the COL3A1 mutation, he did not express the vascular EDS phenotype. This case demonstrates the importance of clinical evaluation of patients carrying the diagnosis of vascular EDS and that successful surgical outcomes may be achieved. The second case depicts the more common clinical course of a patient fully expressing the vascular EDS phenotype. Awareness of differential phenotypic expression of the vascular EDS, aided by knowledge of a patient’s previous response to surgery and trauma, is important in planning elective operative therapy.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) received no financial support for the research, authorship, and/or publication of this article.