
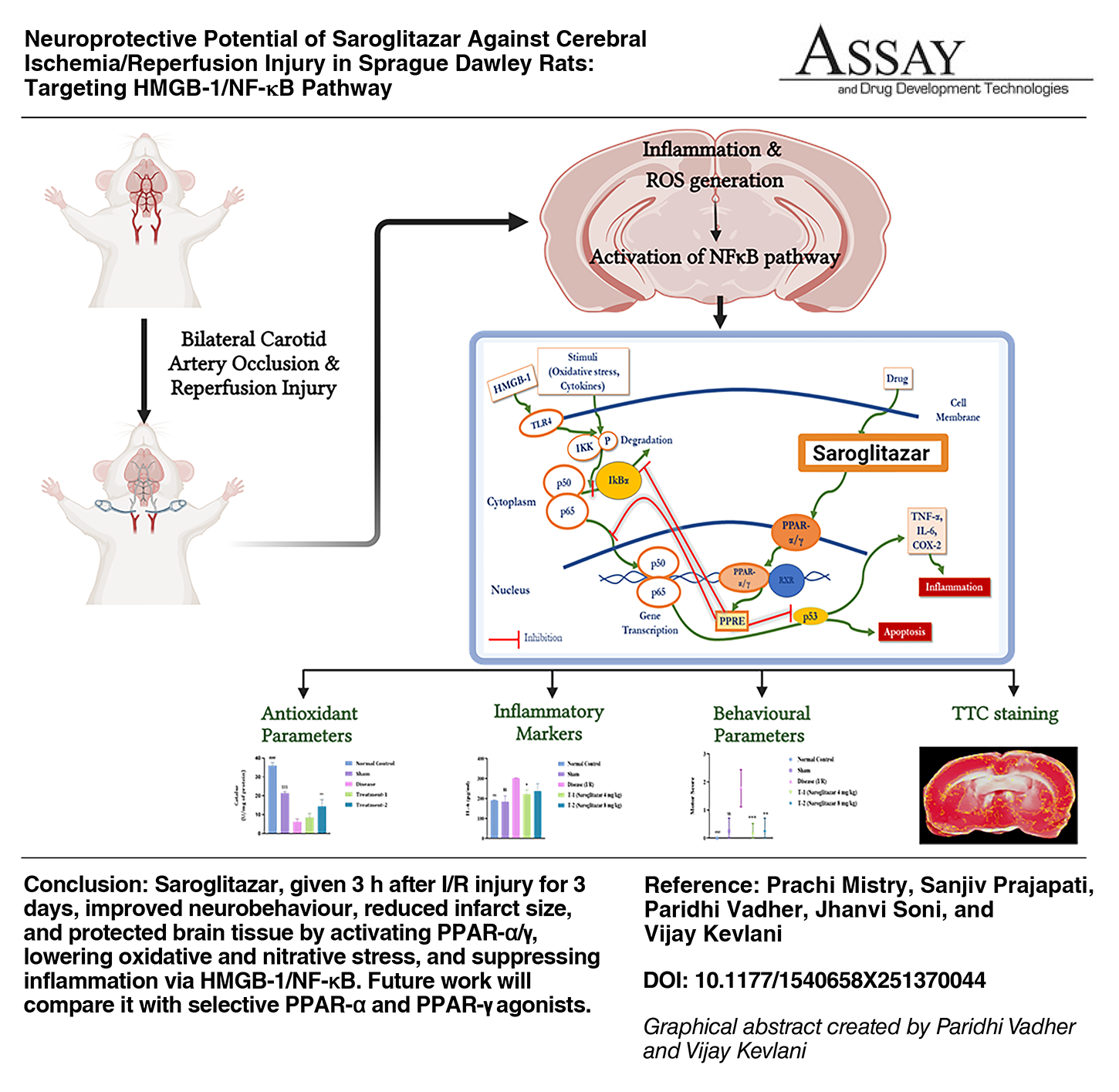
Abstract
Stroke is an intricate oxidative and inflammatory response resulting from cerebral ischemia followed by reperfusion injury. The complex pathophysiology of stroke poses challenges for treatment. Peroxisome proliferated-activated receptor (PPAR) expression in the rat hippocampus is markedly elevated post cerebral ischemia/reperfusion injury (I/R injury). Hence, Saroglitazar, a dual PPAR-α/γ agonist, was investigated against cerebral I/R injury in rats. Male Sprague Dawley rats were subjected to bilateral common carotid artery occlusion for 30 min and reperfusion for 3 days. During the reperfusion, animals were treated with vehicle or Saroglitazar once a day for 3 days. The behavioral parameters were assessed, and animals were sacrificed to measure oxidative markers (Malondialdehyde, Superoxide dismutase, catalase, and reduced glutathione), inflammatory markers (interleukin-6, tumor necrosis factor-α, nuclear factor kappa-light chain enhancer of activated B cells [NF-κB], and high mobility group box 1 protein [HMGB-1]), infarction, and histopathology changes. Following I/R injury, antioxidant enzymes were reduced, while nitric oxide and inflammatory markers were increased in the disease group. In the rat hippocampus, these changes led to neurobehavioral impairment and cerebral infarction. Saroglitazar improved the levels of antioxidants and reduced inflammation. 2,3,5-triphenyltetrazolium chloride staining and histopathological analysis revealed the neuroprotective effect of Saroglitazar in the hippocampus region. The neuroprotective effects of Saroglitazar were attributed to its activation of both PPAR-α and PPAR-γ. It improved antioxidant levels and inhibited pro-inflammatory cytokines by suppressing the HMGB-1/NF-κB signaling pathway. These findings underscore the potential of Saroglitazar in mitigating cerebral I/R injury.
INTRODUCTION
Stroke, a complex oxidative and inflammatory response, is the leading cause of disability and death across the globe. 1 With an annual incidence of 7.59 million and a prevalence of 68.16 million, it stands as a significant health concern. 2 The complex and overlapping pathophysiology of stroke, characterized by oxidative stress, excitotoxicity, blood–brain barrier (BBB) damage, inflammation, and necrosis, 3 presents challenges for treatment. Current therapies fail to adequately address the oxidative and inflammatory damage associated with cerebral ischemia/reperfusion (I/R) injury. 4
Various treatments, such as intravenous thrombolysis and endovascular thrombectomy, have been implemented in the treatment of stroke. The time frame for thrombectomy is 6 h, while for thrombolysis it is 4.5 h, both of which carry the risk of hemorrhagic change. Owing to contraindication, only a limited number of stroke patients have undergone thrombolysis, and approximately 50% of these individuals achieved complete recovery. Moreover, both thrombolysis and thrombectomy lack neuroprotection and have restricted practical applicability. Thus, there is a necessity for neuroprotective drugs to mitigate cerebral lesions and enhance functional outcomes. 5
The nuclear factor kappa-light chain enhancer of activated B cells (NF-κB) and high mobility group box 1 (HMGB-1) protein exacerbate cerebral I/R injury. 6 HMGB-1, a pro-inflammatory protein, stimulates toll-like receptor 2 (TLR-2) and TLR-4, resulting in the activation of NF-κB. HMGB-1 also promotes the receptor for advanced glycation end product (RAGE), stimulating mitogen-activated protein kinases (MAPKs), leading to NF-κB activation. NF-κB triggers pro-inflammatory genes, including inducible nitric oxide synthase (iNOS), tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6), leading to inflammation. 7
Peroxisome proliferator-activated receptors (PPARs) are ligand-inducible nuclear transcription factors, governing the expression of genes related to lipogenesis, lipolysis, and metabolic homeostasis. 8 The PPAR family consists of three receptors: PPAR-α, PPAR-β, and PPAR-γ. 9 After I/R damage, PPARs expression in the rat hippocampus is markedly elevated, culminating at 24 h for PPAR-α and PPAR-γ and 48 h for PPAR-β. 10 Fenofibrate, a PPAR-α agonist, enhances neurogenesis and diminishes inflammation in the hippocampus by inhibiting NF-κB and MAPK pathways. 11 Fenofibrate inhibits the expression of HMGB-1. 12 The PPAR-γ agonist, Rosiglitazone, protects against inflammation and apoptosis by reducing HMGB-1/RAGE expression. 13 It safeguards the hippocampus against oxidative stress and inflammation by inhibiting the NF-κB pathway in cerebral I/R injury. 14
Saroglitazar, a dual PPAR-α/γ agonist, decreases inflammatory markers and enhances antioxidant biomarkers, providing protection against Parkinson’s disease, 15 diabetic dyslipidemia, 16 renal fibrosis, 17 epilepsy, 18 and nonalcoholic fatty liver disease. 19 Saroglitazar substantially alleviates neurotoxicity caused by 3-nitropropionic acid by suppressing cholinesterase enzymes, reducing oxidative stress and neuroinflammation. It improves memory skills and adult neurogenesis through the activation of Wnt β-catenin signaling in dementia. 20 Saroglitazar mitigated Alzheimer’s disease by inhibiting scopolamine-induced learning and memory impairments, oxidative stress, and cholinergic injury. 21 Considering the neuroprotective properties of PPAR-α and PPAR-γ agonists, we propose that Saroglitazar may similarly confer advantages in cerebral I/R injury. This work aims to investigate the antioxidant and anti-inflammatory properties of Saroglitazar, emphasizing the involvement of the NF-κB and HMGB-1 signaling pathways in neuroprotection.
MATERIALS AND METHODS
Materials
Saroglitazar was received as a gift sample from Zydus Research Centre, Ahmedabad, India. Ketamine injection IP (Claris Life Science Ltd., Gujarat, India), Midazolam injection IP (Claris Life Science Ltd., Gujarat, India); 2,3,5-triphenyltetrazolium chloride (TTC), 2-thiobarbituric acid (TBA), L-adrenaline (Lobs Chemicals, India); 1,1,3,3-tetraethoxy propane (TEP), 5,5′-dithiobis-(2-nitrobenzoic acid) (Ellaman’s reagent) (Sigma Aldrich, CA, USA); Folin-Ciocalteus phenol reagent, crystalline bovine serum albumin (BSA), N-(1-naphthyl) ethylenediamine dihydrochloride (BMR reagent) (Sisco Research Laboratories, India); diclofenac sodium (Troika Pharmaceuticals Ltd., India); and gentamycin (Procyon Life Science, India) were procured commercially.
Experimental Animals
Overall, 45 male Sprague Dawley rats (10–12 weeks old, 250–280 g) were procured from Syncbio Research Pvt. Ltd., Gujarat, India, and housed at the animal house of L.J. Institute of Pharmacy, Ahmedabad, India. Animal care was given as per Committee for Control and Supervision of Experiments on Animals (CCSEA) guidelines, maintaining a 25 ± 2°C temperature, 40%–70% relative humidity, a 12-h light/dark cycle, and free access to water. We acclimatized the rats for 1 week prior to the experiments. The Institutional Animal Ethics Committee (IAEC) at the L.J. Institute of Pharmacy, Ahmedabad, approved the protocol under protocol no. LJIP/IAEC/2023-24/01.
Induction of I/R Injury and Treatment Groups
Transient global cerebral ischemia was induced by bilateral common carotid artery (BCCAO) occlusion. Animals were divided into five groups (n = 9) as follows:< id="332" data-dummy="list" list-type="normal">
Group I (normal control): 0.5% w/v carboxy methyl cellulose (CMC) (p.o.) Group II (sham): CCA exposed but not occluded, 0.5% w/v CMC (p.o.) Group III (disease): 30 min of CCA occlusion followed by 72 h of reperfusion, 0.5% w/v CMC (p.o.) Group IV (treatment 1: low-dose Saroglitazar): 30 min CCA of occlusion followed by 72 h of reperfusion, 4 mg/kg Saroglitazar (p.o.) for 3 days (once a day) Group V (treatment 2: high-dose Saroglitazar): 30 min CCA of occlusion followed by 72 h of reperfusion, 8 mg/kg Saroglitazar (p.o.) for 3 days (once a day)
</>Rats in groups III, IV, and V were subjected to BCCAO under general anesthesia induced by ketamine (45 mg/kg, i.p.) and midazolam (5 mg/kg, i.p.). The anesthesia was confirmed by the absence of reflexive responses to aversive stimuli. Animals were placed on their back, and their paws were fixed to the pad using adhesive tape. The surgical site was prepared by removal of hair, application of local anesthetic (2% lidocaine), and antibacterial solution (5% povidone-iodine). A midline incision of 2 cm was made on the neck; both the CCAs were carefully separated from the vagus nerve and occluded simultaneously for 30 min with surgical thread. A small tube (2 mm diameter) was inserted between the artery and the thread to avoid arterial damage. 22 Reperfusion was allowed for 72 h postischemia. Saroglitazar (4 and 8 mg/kg) was given during the reperfusion period for 3 days (once a day) to groups IV and V, respectively. The dosage selection of Saroglitazar in our study was supported by prior literature indicating that the 3–10 mg/kg dose exhibited neuroprotective benefits in Alzheimer’s disease and 3-nitropropionic acid-induced neurotoxicity models. The 8 mg/kg dose was chosen to investigate a possible dose-dependent impact and assess if an increased dosage would offer enhanced neuroprotection. The oral mode of administration was selected due to previous studies demonstrating effective systemic bioavailability and neuroprotective effectiveness of Saroglitazar when delivered orally.20,21 Moreover, oral delivery closely resembles the anticipated clinical route, hence augmenting the translational significance of the study. The treatment duration of 3 days was selected based on previous studies showing that the expression of PPAR-α and PPAR-γ peaks at approximately 24–48 h postinjury. Extending the therapy to 3 days was intended to provide sustained receptor engagement and modulation beyond the initial peak. This additional exposure ensures that the therapeutic effects of Saroglitazar continue during the critical early phase of injury, potentially enhancing neuroprotection by covering both the peak and the subsequent phase of receptor activity.10,23 Diclofenac sodium (6.75 mg/kg, i.m.) and gentamicin (40 mg/kg, i.m.) were used to manage the pain and to avoid infection, respectively. Group II animals underwent the same procedure without BCCA occlusion (Fig. 1).
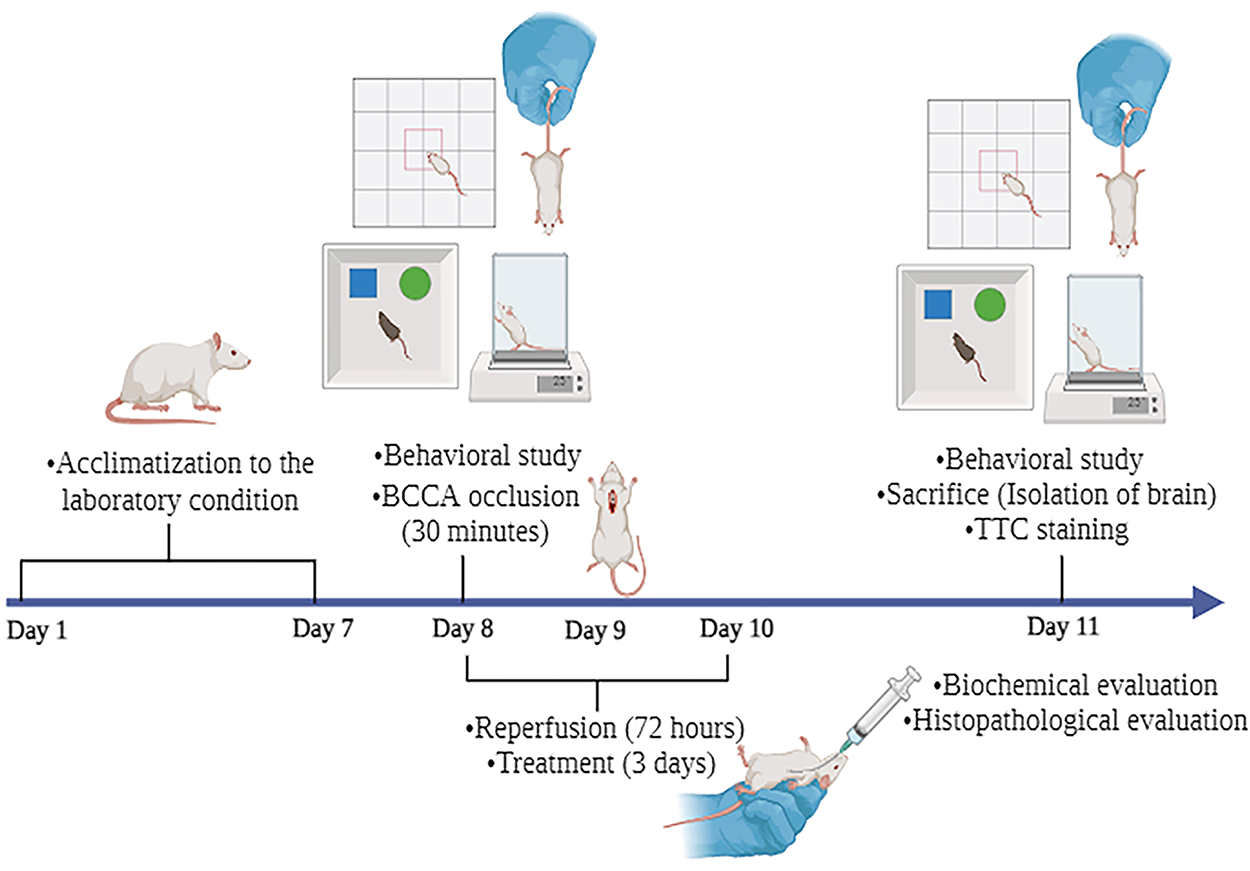
Schematic representation of experimental protocol. After a 7-day acclimatization period, the animals were divided into five groups (n = 9): Group I (normal control), Group II (sham), Group III (disease), Group IV (treatment 1), and Group V (treatment 2). Behavioral studies were performed on day 8 (preischemia) and 11 (post-72-h-reperfusion). On day 11, the animals were sacrificed, and their brains were isolated for biochemical analysis, infarct area measurement, and histopathological evaluation.
Preparation of Postmitochondrial Supernatant
After 72 h of reperfusion, rats were sacrificed under ketamine overdose, followed by cervical dislocation. The rats were dissected, brains were collected, rinsed in cold 0.9% saline, blotted dry, and weighed. A 10% (w/v) homogenate was immediately prepared in cold phosphate-buffered saline (0.05 M, pH 7.4) using a homogenizer. The homogenate was centrifuged at 10,000 g for 10 min at 4°C (NEYA 16 R, Remi Elektrotechnik Ltd.) to obtain the postmitochondrial supernatant and stored at −20°C until use. 22
Biochemical Estimation of Oxidative Stress Markers
Malondialdehyde
Malondialdehyde (MDA) levels were estimated using the TBA reaction method. 24 Briefly, 2.0 mL of 10% TCA was added to 1.0 mL of homogenate and centrifuged at 3,000 g for 10 min at room temperature. The supernatant (2.0 mL) was mixed with 0.5 mL of 1% TBA and heated at 95°C for 60 min to produce a pink-colored MDA–TBA complex. After cooling, the absorbance was measured at 532 nm using a molar extinction coefficient of 0.156 × 106 M−1 cm−1.
Superoxide dismutase
Superoxide dismutase (SOD) was determined based on its ability to inhibit the autoxidation of Epinephrine to adrenochrome at alkaline pH. 22 In total, 0.5 mL of 0.3 M carbonate–bicarbonate buffer (pH 10.2) and 0.5 mL of 0.6 mM EDTA were added to the 0.2 mL sample, and the final volume was adjusted to 2.5 mL. Overall, 0.4 mL of 1.8 mM epinephrine was added to the mixture, and the absorbance was measured at 420 nm for 1 min. The specific activity of SOD was expressed as U/mg of protein.
Catalase
Catalase activity was measured by the decomposition of H2O2 and the corresponding decrease in absorbance at 240 nm. Briefly, 1.9 mL of 50 mM phosphate buffer (pH 7.0) and 1.0 mL of 30 mM H2O2 were added to 1.0 mL of homogenate. Absorbance was recorded at 0.0, 1.0, and 2.0 min at 240 nm. Catalase activity was expressed as units/mg of protein/min using a molar extinction coefficient of 43.6 M−1 cm−1. 25
Reduced glutathione
Glutathione (GSH) was estimated to use its reaction with DTNB. 26 In total, 0.1 mL of homogenate, 0.9 mL of 0.1% Na-EDTA, and 1.5 mL of a 20% TCA mixture were allowed to stand (for 5 min) before centrifugation (10 min at 3,000 g). Overall, 1.8 mL of 1 mM DTNB was added to 0.2 mL of supernatant, and absorbance was read at 412 nm. GSH concentration was expressed as mM/mg of protein using a molar extinction coefficient of 1.36 × 104 M−1 cm−1.
Nitric oxide
Nitric oxide (NO) reacts with oxygen to produce nitrate and nitrite. 27 About 5 mL of 0.1% N-(1-naphthyl) ethylenediamine dihydrochloride and 5 mL of 1% sulfanilic acid were mixed to prepare the Griess reagent. Sample solutions were prepared by adding 100 μL of Griess reagent and 2.6 mL of deionized water to 100 μL of homogenate and incubating for 30 min at room temperature. NO concentration was calculated by comparison with a standard sodium nitrite (NaNO2) solution at 543 nm and expressed as μM/mL.
Total thiol
The total thiol assay was based on the reaction of sulphydryl groups with DTNB to form a relatively stable yellow color. 28 Briefly, 0.6 mL of 0.2 M tri-EDTA buffer, 40 mL of 10 mM DTNB, and 3.16 mL of methanol was added to 0.2 mL of homogenate. This mixture was centrifuged (10 min, room temperature), and the absorbance was measured at 412 nm. Total thiol content was expressed as mM/mL using the molar extinction coefficient of 13.6 × 103 M−1 cm−1.
Total protein
The total protein content of brain homogenate (10% w/v) was determined as per the method described before. 29
Estimation of Inflammatory Markers
Neuroinflammatory markers (IL-6, TNF-α, NF-κB, and HMGB-1) in the brain homogenate were measured using a commercially available ELISA kit (Allianz BioInnovation, India) according to the manufacturer’s instructions.
Behavioral Studies
All the behavioral studies were performed by an individual unknown to the experiment.
Novel object recognition test
The novel object recognition test (NORT) was used to assess the tendency of rats to explore novelty (based on their natural preference to spend more time with a novel object than a familiar one), reflecting learning and recognition. 30 The test included two sessions: first, rats explored two identical objects for 2 min; second, one object was replaced with a novel one, and exploration time was recorded. Discrimination toward the novel object indicated recognition. 31
Open field test
The open field test is used to assess locomotor ability and exploration behavior. 32 The apparatus consists of a wooden box (50 cm × 50 cm), with the bottom divided into 36 equal squares (inter zone: 4 squares in the center; outer zone: 32 squares in the periphery). 33 All the animals were allowed to explore the open field for 5 min in a dark environment. Moving time percentage and total squared crossings by the animals were recorded and analyzed as exploratory behavior. Additionally, rearing behavior was also analyzed. After each animal test, the apparatus was cleaned with 75% alcohol and water to avoid any interference due to smell. 34
Motor test
In the motor test, each parameter was scored from 0 (normal) to 3 (maximum). When raising a rat by the tail, flexion of the forelimb (1), flexion of the hindlimb (1), and movement of the head more than 10° to the vertical axis within 30 s (1) was measured. Walking was scored as follows: normal (0), inability to walk straight (1), circling toward the paretic side (2), and falling to the paretic side (3). 35
Neurological score (Bederson score)
Neurological score was used to evaluate neurological deficit based on modified Bederson score. 36 Scoring was done according to the following criteria: extension of both forelimbs (0); forelimb hemiparesis while held by the tail (1); cortex damage indicated by circling toward the paretic side (2); striatal damage indicated by no spontaneous movement (3); and extensive brain damage indicated by animal death (4).
Measurement of Infarction Area
Seventy-two hours after reperfusion, rats were sacrificed; brains were isolated, frozen, and cut into 2–3 mm thick coronal slices. Each slice was immersed in 1% TTC for 15 min at 37°C, then fixed in a 10% buffered formalin solution overnight. 37 The infarct area was measured using ImageJ.
Histopathology
The selected brains from each group were fixed with 10% formalin overnight, embedded in paraffin wax, and cut into a longitudinal section of 5 μm thickness. From each brain, three sections were collected. The sections were stained with hematoxylin and eosin dye for histopathological analysis.
Statistical Analysis
All the values are expressed as mean ± standard error of mean. 38 Statistical analysis was performed using one-way analysis of variance, 38 followed by Dunnett’s test for comparison between multiple groups. A Kruskal–Wallis (nonparametric) test was performed to compare the differences in scores between multiple groups. Statistical analysis was done using GraphPad Prism (Version 9, GraphPad software, California, USA); p < 0.05 was considered to be statistically significant.
RESULTS
Effect of Saroglitazar Against Oxidative Stress Induced by Cerebral I/R Injury
Cerebral I/R injury significantly decreased antioxidant levels (SOD, catalase, and GSH) in the disease group compared to the sham group. MDA, a key biomarker of membrane damage, was significantly elevated post-I/R injury in the disease group (22.10 ± 0.11, #p < 0.05) compared to the sham (16.14 ± 0.14). High-dose Saroglitazar (18.47 ± 0.70, *p < 0.05) decreased MDA significantly compared to the disease group (Fig. 2A).
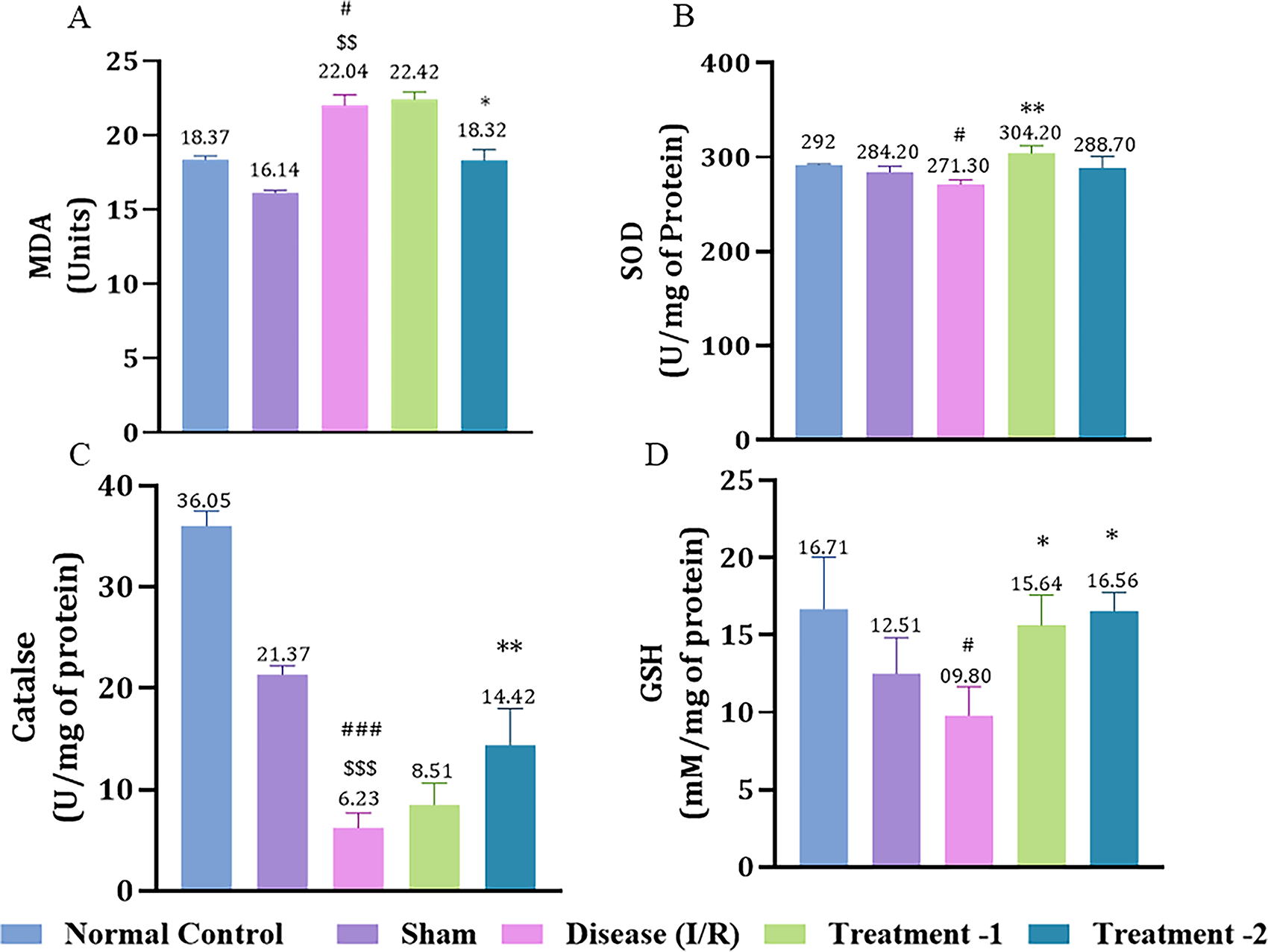
Effect of Saroglitazar on
SOD was reduced in the disease group (271.3 ± 3.51, #p < 0.05) compared to the normal control (292.00 ± 0.49). Saroglitazar markedly elevated SOD level at a lower dose (304.2 ± 4.52, **p < 0.01) compared to the disease (Fig. 2B). Similarly, catalase activity was markedly diminished in the disease group (6.24 ± 0.74, ###p < 0.001) compared to the sham (21.37 ± 0.63). High-dose Saroglitazar significantly elevated catalase. GSH levels were significantly depleted in the disease control group (9.80 ± 1.30, #p < 0.05) compared to the normal control group (16.34 ± 1.02). In contrast, Saroglitazar treatment led to a significant increase in GSH levels at both the lower dose (17.38 ± 0.95, *p < 0.05) and the higher dose (16.56 ± 0.70, *p < 0.05), compared to the disease control group, highlighting its potential to restore antioxidant capacity. These findings collectively demonstrate the antioxidant effects of Saroglitazar in a dose-dependent manner, as evidenced by the restoration of SOD, catalase, and GSH levels following cerebral I/R injury.
Effect of Saroglitazar on NO Level in Rats Exposed to Cerebral I/R Injury
I/R injury significantly elevated NO levels in the disease group (24.71 ± 0.22, #p < 0.05) compared to the sham (21.27 ± 0.33) group. High-dose Saroglitazar attenuated nitrative damage (21.27 ± 1.00, *p < 0.05) in I/R injured rats (Fig. 3A).
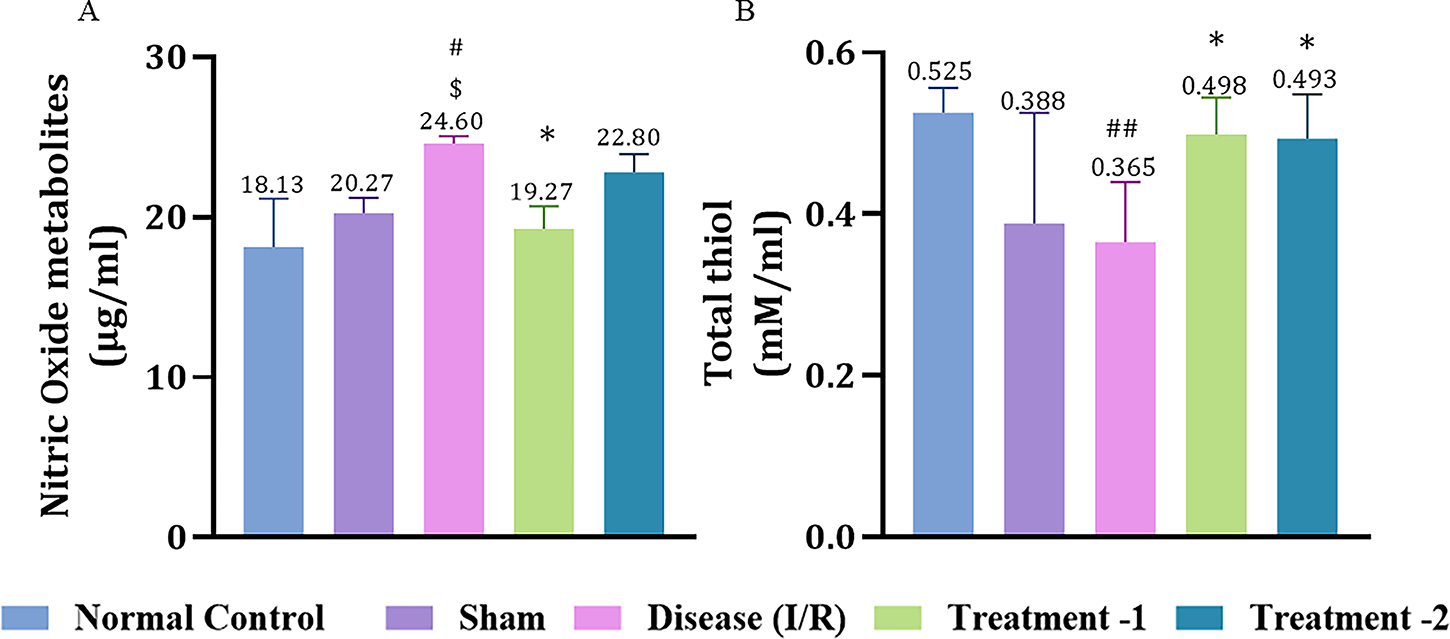
Effect of Saroglitazar on NO and total thiol levels in rats exposed to cerebral I/R injury. Animals were treated with different doses of Saroglitazar (4 and 8 mg/kg, p.o.) for 3 days. Data are represented as mean ± SEM. Statistical analysis was performed by one-way ANOVA and post hoc Dunnett’s test, where n = 6, p < 0.05. (*p < 0.05 vs. disease, #p < 0.05 vs. normal control, ##p < 0.01 vs. normal control, $p < 0.05 vs. sham.) IR, ischemia/reperfusion; NO, nitric oxide.
Effect of Saroglitazar on Total Thiol Level in Rats Exposed to I/R Injury
Rats in the disease group showed a significantly reduced total thiol level (0.37 ± 0.03, ##p < 0.01) compared to the normal control (0.52 ± 0.01). Both high- and low-dose Saroglitazar significantly increased total thiol levels (0.48 ± 0.01, 0.47 ± 0.01, *p < 0.05, respectively) compared to the disease group (Fig. 3B).
Effect of Saroglitazar on HMGB-1, TNF-α, IL-6, and NF-κB Expression in Rats Exposed to I/R Injury
IL-6 expression was significantly higher in the disease group (302.7 ± 1.333, **p < 0.01) compared to the sham (184.3 ± 20.33). Low-dose Saroglitazar restored (221.8 ± 10.90, *p < 0.05) the elevated IL-6 expression after an I/R injury compared to the disease group (Fig. 4A). I/R injury significantly raised TNF-α (233.6 ± 10.88, #p < 0.05) compared to the sham group (170.4 ± 8.125). Low-dose Saroglitazar markedly reduced TNF-α (175.7 ± 6.858, **p < 0.01) compared to the disease group (Fig. 4B).
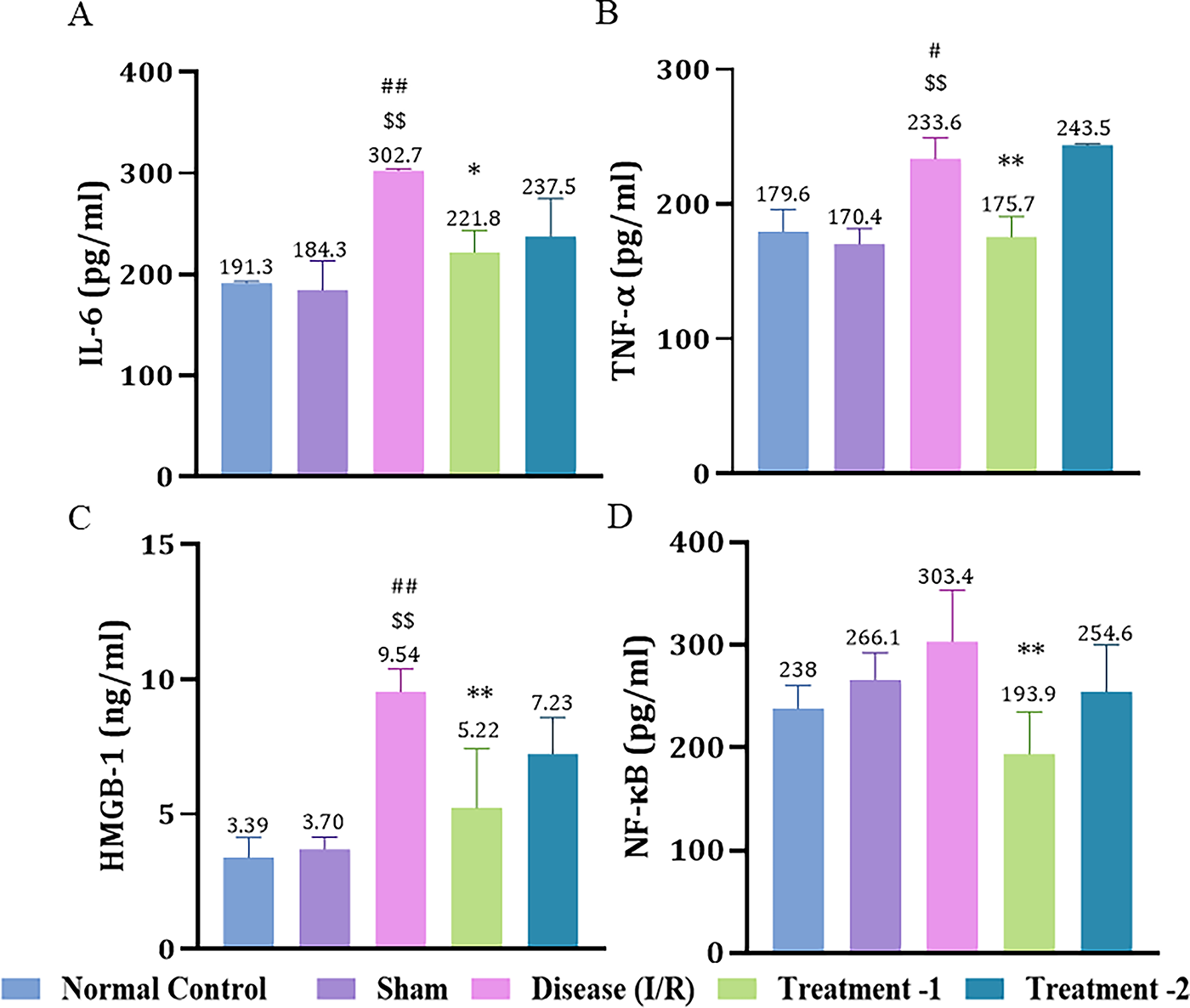
Effect of Saroglitazar on
The expression of HMGB-1 was markedly increased in the disease group (9.54 ± 0.49, ##p < 0.01) compared to the sham (3.70 ± 0.32) group. Low-dose Saroglitazar effectively reduced (5.23 ± 0.90, **p < 0.01) HMGB-1 expression as compared to the disease group (Fig. 4C). There was a significant difference in NF-κB expression after low-dose Saroglitazar (193.9 ± 18.30, **p < 0.01) compared to the disease group (Fig. 4D).
Effect of Saroglitazar on NORT in Rats Exposed to I/R Injury
I/R injury significantly impaired recognition memory in the disease group (0.27 ± 0.04, ##p < 0.01) compared to the sham (0.58 ± 0.04) group. Both low- and high-dose Saroglitazar-treated groups significantly restored recognition memory (0.57 ± 0.05, **p < 0.01; 0.76 ± 0.01, ***p < 0.001; respectively) compared to the disease group (Fig. 5).
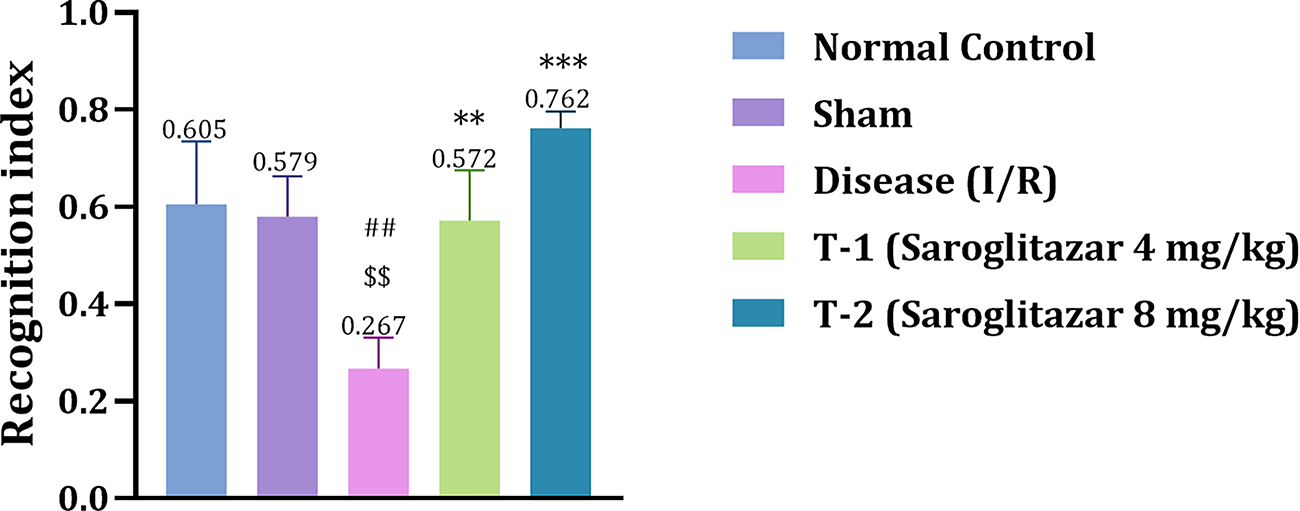
Effect of Saroglitazar on recognition memory (NORT) in rats exposed to cerebral ischemia-reperfusion injury. Animals were treated with different doses of Saroglitazar (4 and 8 mg/kg, p.o.) for 3 days. Data are represented as mean ± SEM. Statistical analysis was performed by one-way ANOVA and post hoc Dunnett’s test, where n = 6, p < 0.05. (***p < 0.001 vs. disease, **p < 0.01 vs. disease, ##p < 0.01 vs. normal control, $$p < 0.01 vs. sham.)
Effect of Saroglitazar on an Open Field Test in Rats Exposed to I/R Injury
In the open field test, we measured percentage moving time, number of squares crossed, and rearing behavior. Percentage moving time was significantly reduced in the disease group (28.58 ± 4.199, ###p < 0.001) compared to the sham group (53.86 ± 5.20). Both low- and high-dose Saroglitazar significantly improved percentage moving time (54.73 ± 1.64, *p < 0.05 and 63.39 ± 9.061, **p < 0.01; respectively) compared to the disease group (Fig. 6A). A substantial decrease in the number of squares crossed was noted in the disease group (34.75 ± 8.15, ###p < 0.001) compared to the normal control group (84.75 ± 14.08). High-dose Saroglitazar significantly restored the activity (72.75 ± 1.75, **p < 0.01) compared to the disease group (Fig. 6B). I/R insult in the disease group reduced rearing behavior of rats in the open field apparatus (18.33 ± 2.02, ##p < 0.01) compared to the normal control group (21.00 ± 2.12). Treatment with Saroglitazar did not improve in rearing behavior (Fig. 6C).
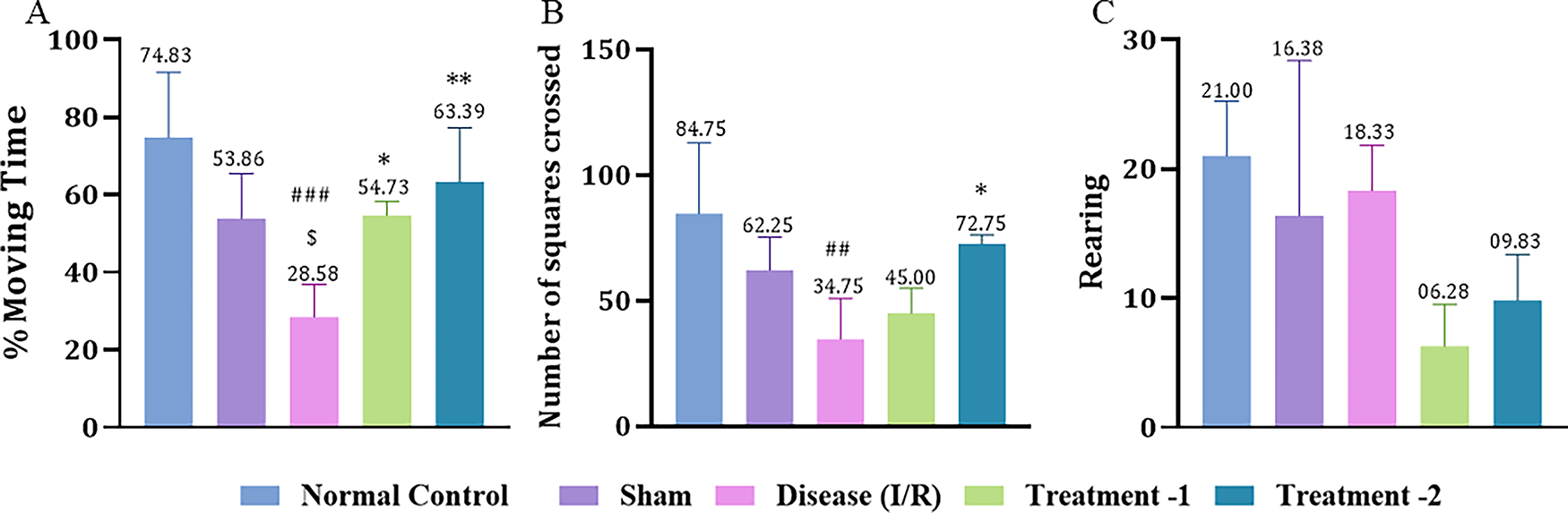
Effect of Saroglitazar on open field activity in rats exposed to cerebral I/R injury. Animals were treated with different doses of Saroglitazar (4 mg/kg and 8 mg/kg, p.o.) for 3 days. Behavior changes were observed by measuring
Effect of Saroglitazar on Motor Test in Rats Exposed to Cerebral I/R Injury
In the motor test, two behaviors were observed: when raised by the tail and when placed on the floor. In the tail raising test, the disease group showed a significant increase in motor score (2.444 ± 0.17, ###p < 0.001) compared to the sham group (0.25 ± 0.16). Saroglitazar low- and high-dose showed a significant reduction in the motor score (0.28 ± 0.18, **p < 0.01 and 0.25 ± 0.16, ***p < 0.05, respectively) compared to the disease group (Fig. 7A).
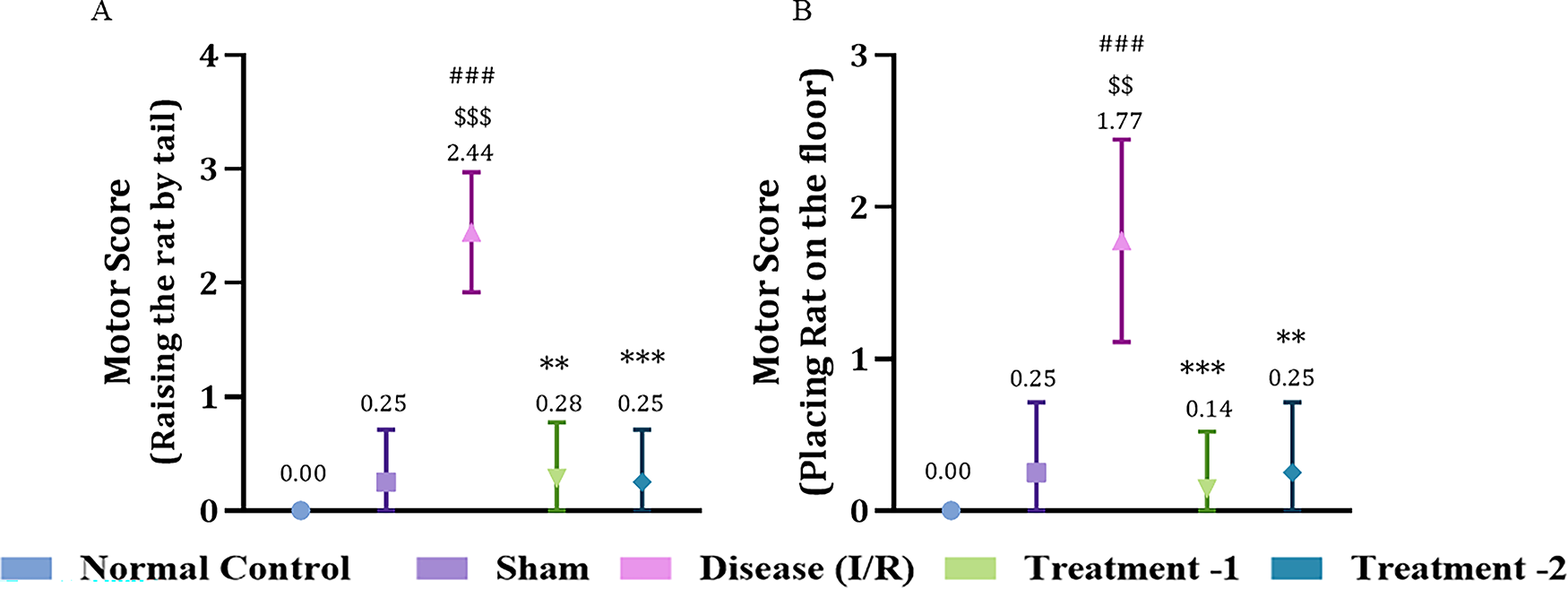
Effect of Saroglitazar on motor activity in rats exposed to cerebral I/R injury using the Kruskal–Wallis test. Representative analysis for motor score when
When placed on the floor, the motor score significantly increased in the disease group (1.78 ± 0.22, ###p < 0.001) compared to the sham group (0.25 ± 0.16). Both low- and high-dose Saroglitazar showed significantly less motor activity (0.1429 ± 0.1429, ***p < 0.001 and 0.25 ± 0.1637, **p < 0.01) compared to the disease group (Fig. 7B).
Effect of Saroglitazar on Neurological Score (Bederson Score) in Rats Exposed to Cerebral I/R Injury
Based on the functional assessment, cerebral ischemia caused neurological impairment. I/R injury in the disease group (2.54 ± 0.25, ###p < 0.001) showed a significant increase in neurological score compared to the sham (0.12 ± 0.12) group. Both low- and high-dose significantly improved functional recovery by reducing neurological score (0.37 ± 0.26, ***p < 0.001 and 0.37 ± 0.26, **p < 0.1, respectively) compared to the disease group (Fig. 8).
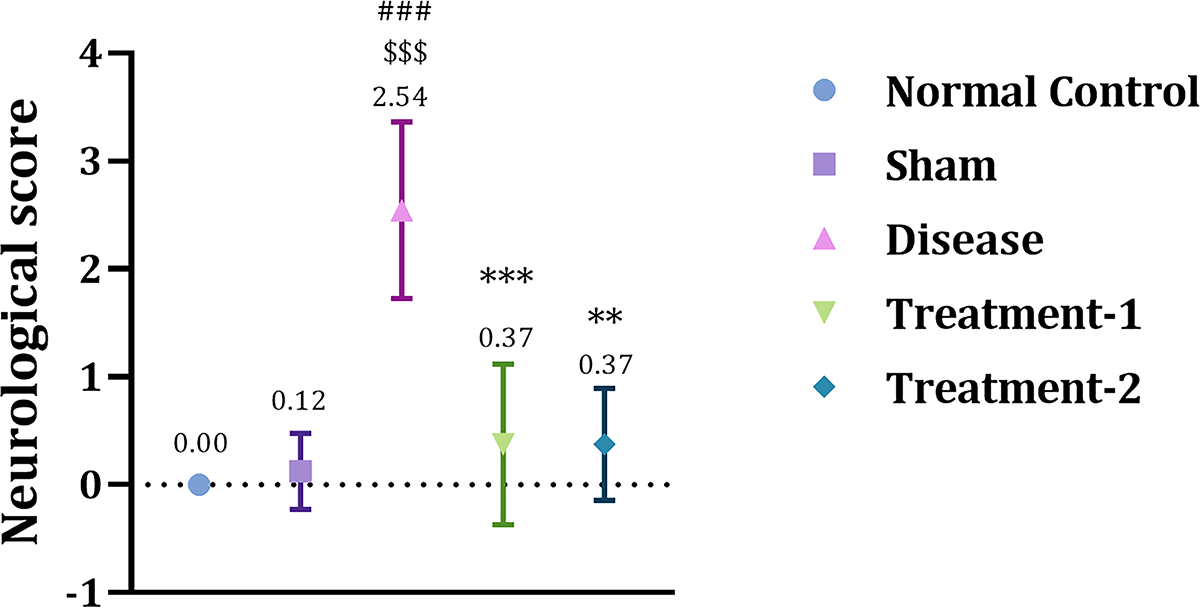
Effect of Saroglitazar on neurological score in rats exposed to cerebral I/R injury using Kruskal–Wallis test. Animals were treated with different doses of Saroglitazar (4 and 8 mg/kg, p.o.) for 3 days. Scoring criteria: extension of both forelimbs (0); forelimb hemiparesis while held by the tail (1); cortex damage indicated by circling toward the paretic side (2); striatal damage indicated by no spontaneous movement (3); extensive brain damage indicated by animal death (4). Data are represented as mean ± SEM. Statistical analysis was performed by the Kruskal–Wallis test (nonparametric), where n = 6, p < 0.05. (***p < 0.001 vs. disease, **p < 0.01 vs. disease, ###p < 0.001 vs. normal control.)
Effect of Saroglitazar on Cerebral Infarction in Rats Exposed to Cerebral I/R Injury
Rat brains were isolated and stained with TTC. The infarct area was measured using Image J. After 30 min of ischemia followed by 72 h of reperfusion, the disease group (50.74 ± 0.73) showed a significantly larger infarction area, involving both cortical and subcortical structures, compared to the sham group (28.69 ± 4.40; 31.68 ± 1.68, respectively). Whereas low- and high-dose Saroglitazar-treated groups (32.09 ± 6.24; 26.14 ± 1.86, respectively) showed predominantly red colored tissue after TTC staining in brain slices, indicating a reduction in the total infarction area as compared to the disease group (Fig. 9).
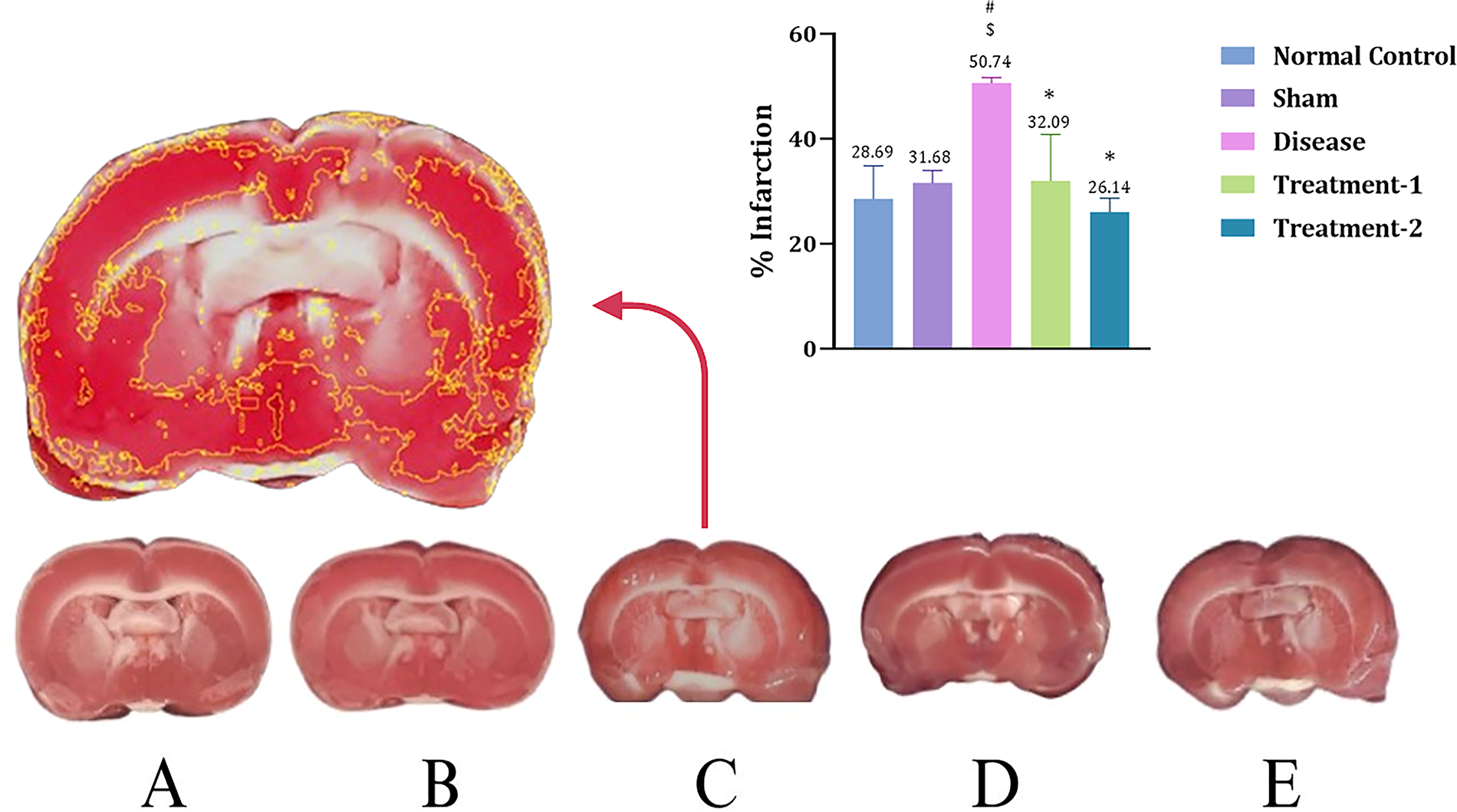
Coronal sections stained with TTC.
Effect of Saroglitazar on Hippocampal Histology in Rats Exposed to I/R Injury
In the present study, H&E staining was performed to observe morphological changes in the hippocampus region of the rat brain (Fig. 10). Coronal sections of the hippocampus from the disease group showed a reduction in granular cell population in the dentate gyrus region compared to the sham group. We also observed congestion and eosinophilic neurons in the hippocampal region of the disease group. Administration of Saroglitazar reduced neuronal degeneration of granular cells and relatively regular morphology compared to the disease group. High-dose Saroglitazar effectively restored histopathological changes in the hippocampus of the rat brain.
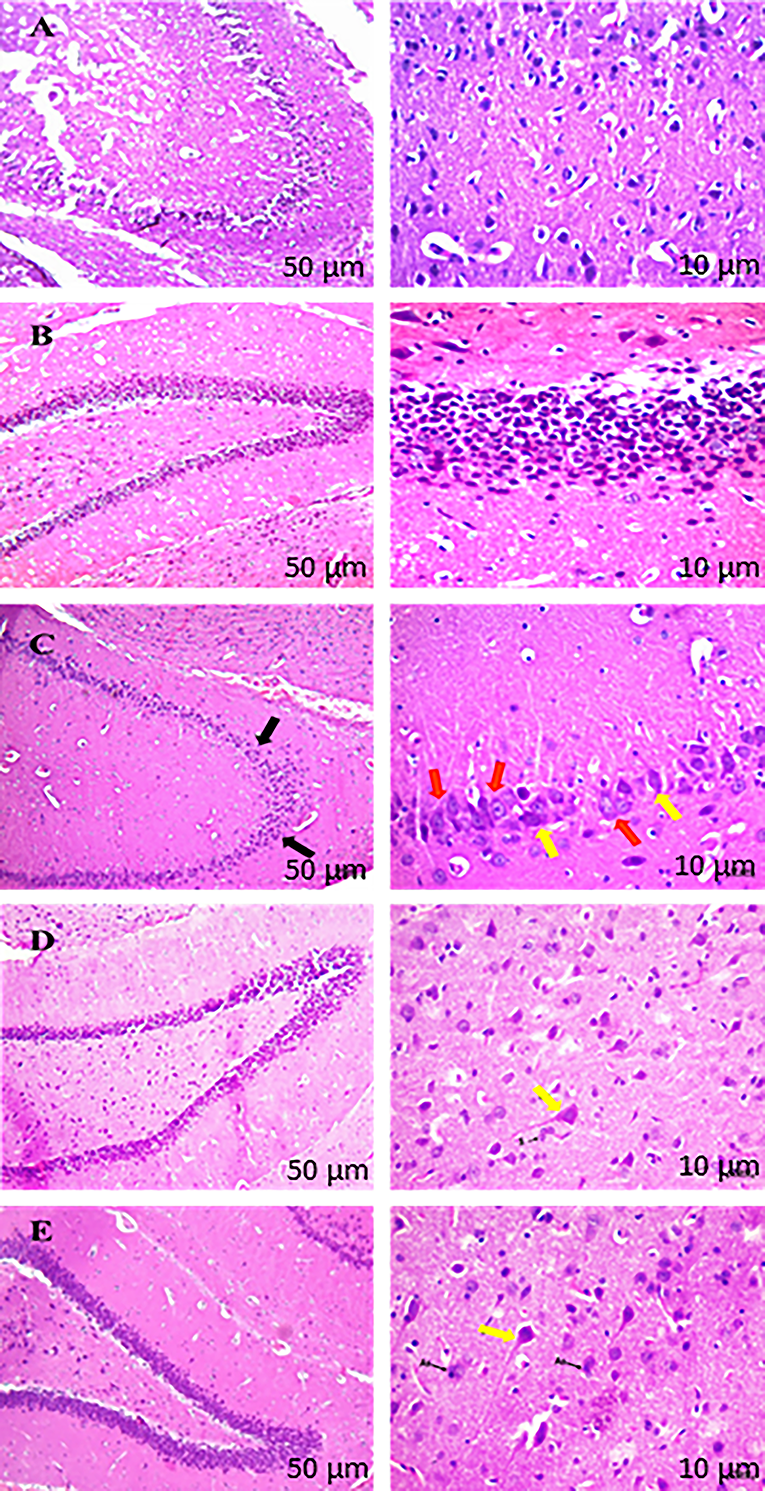
H&E stained the hippocampus region of the rat brain.
DISCUSSION
A stroke transpires when cerebral blood flow is disrupted, resulting in considerable morbidity and mortality. 39 Current therapy fails to fully address the complex pathophysiology of stroke, leading to major clinical challenges. Rehabilitation methods are effective in only about half of stroke patients, underscoring the need for innovative recovery strategies. 40
Oxidative stress and inflammation are critical in the pathophysiology of cerebral I/R injury. Activation of PPARs exerts antioxidant and anti-inflammatory effects via transrepression of transcription factor (NF-κB) and regulation of neuronal death in ischemic conditions. 41 The expression of PPAR-α and PPAR-γ receptors increases within 24 h post-I/R injury, highlighting their role in mitigating I/R insult. 10 This study provides supporting evidence that the dual PPAR agonist, Saroglitazar, may protect the rat brain from transient cerebral I/R injury.
Cerebral I/R injury was induced by BCCAO, which mimics human hypoxic-ischemic injury 42 and results in a 50%–80% reduction in cerebral blood flow, primarily affecting the hippocampus, leading to neuronal damage and deficits in learning and memory. 38 We selected the Sprague Dawley rats for the study, given their high survival rate, satisfactory postoperative recovery, ease of behavioral testing, relatively low costs, and ethical acceptability, making them ideal for investigating cerebral injury.
In both rodent and human models, PPARα and PPARγ exhibit developmentally controlled expression in the central nervous system, with elevated levels during mid-gestation (GD11–13.5 in mice) and markedly reduced levels in adulthood. Notwithstanding this reduction, a modest yet detectable expression of PPARs remains in the adult brain, as evidenced by many molecular approaches (IHC, ISH, RT-PCR). These findings underscore the significance of targeting PPARs in adult central nervous system diseases, including cerebral ischemia/reperfusion damage. 43 Despite variations in expression levels among species, our use of adult Sprague Dawley rats, which possess both PPAR isoforms in the brain, constitutes an appropriate preclinical model. Moreover, Saroglitazar’s pharmacological profile, characterized by its documented antioxidant and anti-inflammatory capabilities, along with previous indications of central nervous system effectiveness in Parkinson’s disease, 15 Alzheimer’s disease, 21 and seizure models, 44 indicates a possibility for brain penetration.
We assessed oxidative stress subsequent to cerebral I/R injury and noted considerable cell membrane damage, indicated by increased MDA levels. The antioxidant enzyme activity was significantly diminished, and NO levels escalated. Furthermore, I/R damage led to increased levels of inflammatory markers, including IL-6, TNF-α, and NF-κB. In the rat hippocampus, these alterations resulted in neurobehavioral impairment, cerebral infarction, and neurological decline. These findings are congruent with prior investigations. 45
Our study demonstrated that cerebral I/R injury significantly elevates lipid peroxidation. 46 Treatment with a low-dose Saroglitazar during the reperfusion period (3 days, once daily) effectively restored MDA levels. This finding aligns with previous research showing that PPAR-α agonist treatment during the reperfusion period decreased MDA levels. 47
Previous human studies have demonstrated that Saroglitazar is effective in improving lipid profiles, particularly by reducing triglyceride levels, and it also exhibits beneficial effects on glycemic control in patients with metabolic disorders. In terms of safety, the clinical data indicate a favorable profile, with most adverse events being mild and transient, such as gastrointestinal discomfort or headache. Overall, the incidence of significant adverse effects is low, supporting its tolerability in clinical use. 48 In previous models of brain injury, particularly those related to neurodegenerative conditions such as Alzheimer’s disease and models of neurotoxicity, Saroglitazar has demonstrated potential benefits by modulating neuroinflammation and oxidative stress through its dual PPAR-α/γ agonist activity.20,21
We observed reduced SOD activity due to cerebral I/R injury in the disease group, though the reduction was not statistically significant. SOD was not significantly reduced in the disease group due to the activation of antioxidant mechanisms in response to oxidative stress from I/R injury. In rats treated with Saroglitazar, SOD levels increased significantly after I/R injury. Previous studies have shown that pretreatment with PPAR-γ agonists like pioglitazone elevated SOD levels in the brain following middle cerebral artery occlusion. 49 The catalase enzyme, crucial in regulating oxidative stress, was significantly reduced after I/R injury. 50 Saroglitazar treatment resulted in a notable increase in catalase expression. This aligns with data indicating enhanced Catalase activity subsequent to PPAR-γ agonist in an intracerebral hemorrhage model. 51 GSH, a key scavenger of reactive oxygen species, was significantly depleted due to I/R injury. 52 Our findings demonstrate that Saroglitazar treatment significantly increased GSH levels in rats post-I/R injury. Similar protective effects against GSH depletion have been observed with PPAR-γ agonists such as rosiglitazone and pioglitazone. 53
Mitochondria, particularly complexes I and III of the respiratory chain, are major sources of oxidative stress. 54 PPAR-γ agonists exert direct effects on mitochondrial respiration, inhibiting both complex I 55 and complex III. 56 Being a PPAR dual agonist, Saroglitazar may have partially disrupted the mitochondrial respiratory chain, influencing both electron transport and superoxide anion production, which may contribute to a decrease in reactive oxygen species generated after I/R injury, resulting in elevated antioxidant enzyme levels after 3 days of treatment.
Due to the I/R injury, excessive production of NO and peroxynitrite (ONOO−), key mediators of neurotoxicity and BBB disruption, has been observed. 57 Saroglitazar significantly reduced NO levels and mitigated ONOO−-induced damage, consistent with findings that the PPAR-α agonist WY14643 reduced NO during cerebral I/R injury by inhibiting inducible iNOS. 58 Thus, Saroglitazar may protect against nitrative stress by inhibiting iNOS during I/R injury. We measured total thiol levels during I/R injury to assess nonenzymatic antioxidant capacity. I/R injury moderately reduced thiol levels, which were restored by Saroglitazar treatment. This aligns with previous findings where Fenofibrate (a PPAR-α agonist) enhanced antioxidant enzyme expression in brain tissue. 59
IL-6 plays a dual role in brain injury, functioning as both a pro- and anti-inflammatory agent. 60 It promotes neuronal apoptosis, inflammatory cytokine release, and compromises BBB integrity. 61 In this study, cerebral I/R injury elevated IL-6 levels, while Saroglitazar treatment moderately reduced IL-6. Similarly, pioglitazone has been shown to protect against traumatic brain injury by activating PPARγ and reducing IL-6 levels. 62 TNF-α, a key cytokine in the inflammatory response during cerebral ischemia, contributes to cell necrosis. 63 In our study, TNF-α levels significantly increased in rats after 72 h of I/R injury. However, Saroglitazar treatment for 3 days effectively reduced TNF-α expression, demonstrating anti-inflammatory effects. This aligns with findings that PPAR-γ activation inhibits TNF-α expression following 3 h of MCAO in rats. 64
HMGB-1, a pro-inflammatory mediator, is released from damaged or necrotic tissues 65 and peaks at 72 h postreperfusion, with prolonged activation. 66 Its pro-inflammatory effect arises from the translocation of HMGB-1 to the cytoplasm and its interaction with RAGE, which activates the MAPK and NF-κB/p53 pathways, triggering cytokine production. 7 Our study revealed a significant increase in HMGB-1 expression after 72 h of reperfusion injury, which Saroglitazar effectively reduced. This aligns with prior research showing pioglitazone mitigates ischemic injury by inhibiting the HMGB-1/RAGE and HMGB-1/TLR-4/NF-κB pathways. 67
NF-κB is a key regulator of pro-inflammatory and apoptosis-related genes. 68 Activated by oxidative stressors like H2O2, ROS, TNF-α, and IL-1β, NF-κB plays a role in the pathophysiology of ischemic stroke. 69 In this study, I/R injury did not significantly elevate NF-κB expression after 72 h, consistent with prior findings that TLR-4/NF-κB expression peaks at 24 h and declines by 72 h postreperfusion. 70 Saroglitazar significantly reduced NF-κB expression, corroborating evidence that pioglitazone decreases NF-κB nuclear translocation in ischemic rats. 14 During I/R injury, HMGB-1 is released, activating NF-κB and promoting the production of pro-inflammatory cytokines like TNF-α and IL-6. 71 Saroglitazar may have reduced IL-6 and TNF-α by directly inhibiting NF-κB expression and indirectly suppressing NF-κB through HMGB-1 blockade.
Oxidative stress and inflammation generated by hypoperfusion are significant factors in neuronal degeneration, resulting in cognitive deficits. 72 Post-I/R injury, we noted a substantial decline in novel object identification scores, signifying diminished spatial memory; a decrease in locomotion time, reflecting a loss of exploratory behavior; an elevation in motor scores, indicating hemiplegia; and an increase in Bederson scores, denoting neurological impairment. Saroglitazar treatment enhanced recognition memory demonstrated a percentage increase in moving time but not in rearing, and improved scores in hemiplegia. It also showed improvement in the Bederson score. Saroglitazar decreased oxidative damage and inflammation following 72 h of reperfusion, which may have improved nociceptive and sensory-motor functioning.
TTC staining is a critical method for assessing cerebral infarct size in rats. 73 In our study, Saroglitazar significantly reduced cerebral infarction after 72 h of I/R injury, consistent with previous findings that pioglitazone reduces infarction and improves neurological deficits via the HMGB-1/RAGE pathway. 67 Post-I/R injury notably impacted the hippocampal dentate gyrus, leading to reduced neurogenesis and impaired nerve regeneration. 74 Saroglitazar provided neuroprotection by restoring hippocampal histopathology, corroborating earlier studies where Rosiglitazone (PPAR-γ agonist) mitigated hippocampal neuronal damage by inhibiting oxidative stress and inflammatory cytokine release. 14
CONCLUSION
Our study demonstrates that oral administration of Saroglitazar, post-3-h I/R injury and continued for 3 days once daily, effectively attenuated neurobehavioral deficits, reduced infarct volume, and restored histopathological integrity. The neuroprotective effects of Saroglitazar are largely attributed to its activation of both PPAR-α and PPAR-γ, which significantly reduced oxidative stress, as evidenced by decreased MDA levels and increased activities of SOD, Catalase, and GSH. Additionally, Saroglitazar was found to reduce nitrative stress. Saroglitazar significantly reduces the expression of pro-inflammatory cytokines, including TNF-α and IL-6, by suppressing the HMGB-1/NF-κB signaling pathway in a PPAR-γ-dependent manner, further enhancing its neuroprotective properties. These findings underscore the therapeutic potential of Saroglitazar in mitigating ischemic brain injury and highlight its dual role in regulating oxidative and inflammatory pathways. One limitation of the study is that apoptosis and microglial involvement were not measured, which could provide further insight into the neuroprotective mechanisms of Saroglitazar. In future prospective studies, the effects of Saroglitazar with those of Fenofibrate, a PPAR-α agonist, and Rosiglitazone, a PPAR-γ agonist, will be compared. This comparative approach will help elucidate the specific contributions of each receptor pathway and further refine therapeutic strategies for brain injury.
Footnotes
ACKNOWLEDGMENTS
The authors extend their sincere gratitude to Dr. Sachin Parmar, Dr. Tejas Sharma, Dr. Vishal Airao, Dr. Prakruti Buch, and Ms. Hital Shah for their invaluable support in developing the research design and imparting essential methodological expertise. Their guidance was pivotal to the success of this study. The authors would also like to thank the L.J. Institute of Pharmacy for providing the research lab to conduct the study.
FUNDING INFORMATION
This work was supported by L.J. Institute of Engineering & Technology, LJK University, under the aegis of NewGen IEDC.
AUTHORS’ CONTRIBUTIONS
P.M.: Conceptualization, formal analysis, investigation, data curation, and writing—original draft. S.P.: Investigation and resources. P.V.: Visualization, writing—original draft, and writing—review and editing. J.S.: Methodology, validation, visualization, supervision, and funding acquisition. V.K.: Conceptualization, methodology, formal analysis, writing—review and editing, resources, funding acquisition, and project administration.
DISCLOSURE STATEMENT
The authors have no relevant financial or nonfinancial interests to disclose.