
Abstract
Antimicrobial resistance (AMR) in Escherichia coli, driven by biofilm formation and quorum sensing (QS), presents a significant challenge in combating infections, particularly urinary tract infections. This study explored the potential of plant bioactive compounds to inhibit LsrR, a key transcriptional regulator of the QS system, in E. coli. The active site of LsrR was identified using the Sitemap module, which demonstrated high druggability, with a D-score of 0.987. Structure-based virtual screening was used to identify plant-derived inhibitors, followed by docking, binding free energy calculations, and induced-fit docking to evaluate ligand interactions and stability. Chebulinic acid, rutin, and vicine have emerged as potent inhibitors with better docking scores and multiple protein–ligand interactions. Molecular dynamics simulations confirmed the stability of these complexes, highlighting their potential to disrupt QS pathways and inhibit bacterial biofilm formation. These findings suggest that plant bioactive compounds are promising novel therapeutic agents for mitigating AMR in E. coli by targeting LsrR.
Keywords
INTRODUCTION
Antimicrobial resistance (AMR) in Escherichia coli is an increasingly urgent problem that affects both human and animal health worldwide. 1 E. coli is a common bacterium found in the intestines of humans and animals, where it typically coexists harmlessly. However, some strains can cause serious infections such as urinary tract infections (UITs), sepsis, and diarrhea. 2 E. coli biofilms are highly organized bacterial communities encased in an extracellular polymeric substance matrix that provides protection from environmental stressors and antimicrobial agents. 3 Biofilm formation is a stepwise process, beginning with reversible surface attachment, progressing to irreversible adherence aided by fimbriae and curli, maturing into a three-dimensional structure, and culminating in the dispersal of cells to initiate new biofilms ( Fig. 1 ).4,5 Central to this process is quorum sensing (QS), a cell density-dependent signaling mechanism that enables bacterial cells to communicate and coordinate behavior. In E. coli, QS is mediated by autoinducers, such as autoinducer-2 (AI-2), which modulate gene expression related to biofilm maturation, virulence factor production, and stress resistance. 6 AI-2 signaling enhances the production of adhesive structures and extracellular components that stabilize biofilms and foster genetic exchanges within the bacterial community, further contributing to antibiotic resistance.7,8
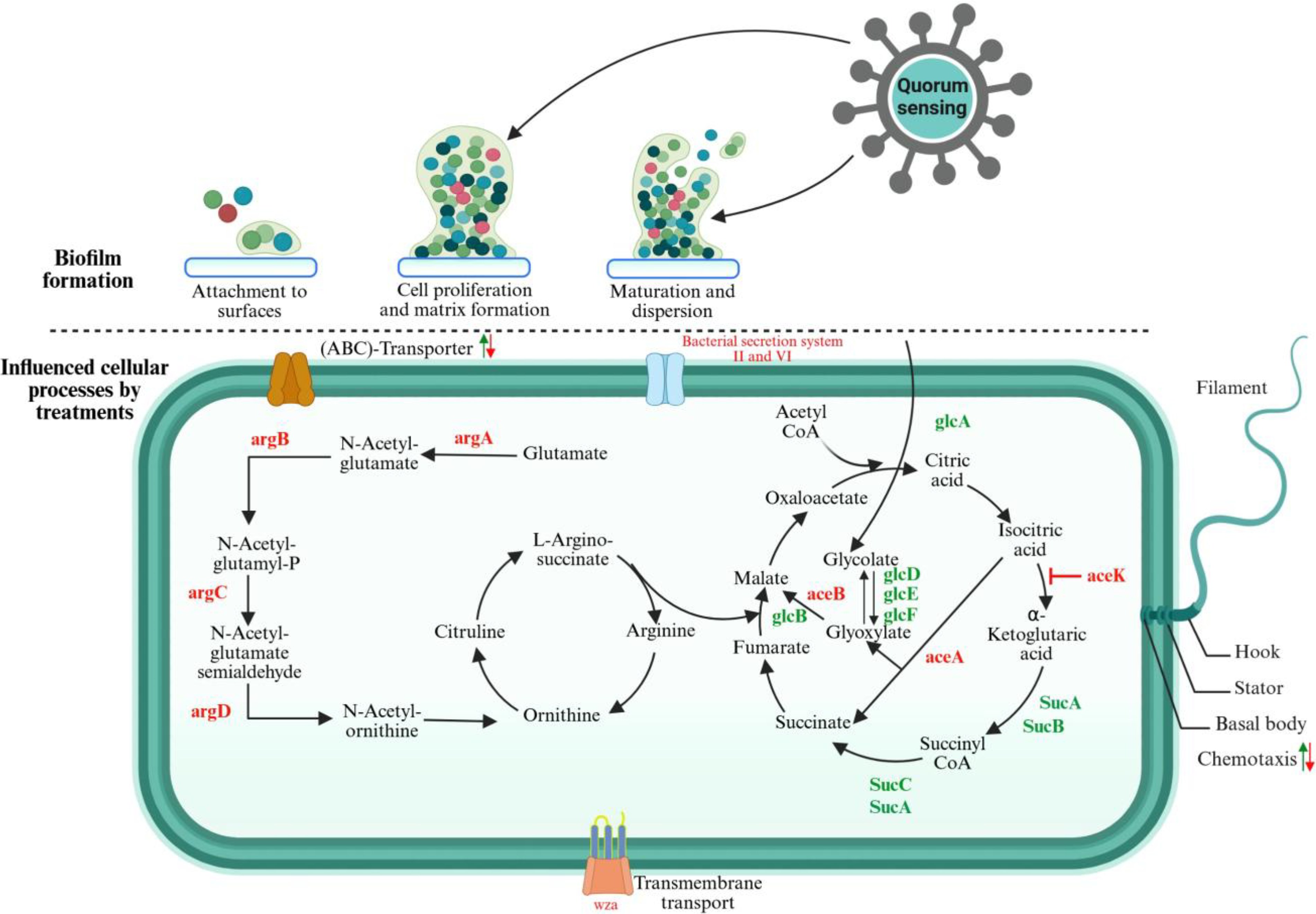
The upper section illustrates the schematic representation of biofilm formation in Escherichia coli, while the lower section delineates the general stages of biofilm development. It also details the signaling pathways and gene expression following the treatment of E. coli biofilm formation. An upward arrow signifies upregulation, whereas a downward arrow denotes downregulation. In certain instances, both regulatory changes were observed upon treatment or in the genes involved. Genes highlighted in green are upregulated, whereas those highlighted in red are downregulated. 4 Created with www.Biorender.com.
Notably, E. coli employs LuxS-like synthase for AI-2 production, which is taken up by the LsrABCD transport system. 9 This QS-regulated pathway governs critical aspects of biofilm physiology, including the activation of stress-resistance genes, modulation of motility, and synthesis of biofilm matrix components.6,10 The QS system also facilitates inter- and intraspecific communication, promoting the establishment of multispecies biofilms in diverse environments. 11 These highly resilient biofilms are implicated in persistent infections, especially in medical device-associated conditions such as catheter-associated urinary tract infections. 12 Understanding the role of QS in E. coli biofilm formation offers promising avenues for developing targeted therapies aimed at disrupting these signaling pathways and potentially mitigating biofilm-mediated infections and antibiotic resistance.
LuxS-regulated repressor (LsrR), a transcriptional regulator, is a central component of QS in E. coli and related bacteria. It acts as a repressor of the lsr (luxS-regulated) operon, which governs the uptake and processing of the signaling molecule AI-2. 13 Upon binding to phospho-AI-2, LsrR undergoes conformational changes that destabilize its tetrameric structure, leading to derepression of QS genes. 14 This regulatory mechanism is critical for controlling bacterial behaviors such as biofilm formation, virulence, and interspecies communication. Structurally, LsrR consists of an N-terminal DNA-binding domain and a C-terminal ligand-binding domain, which together enable its role as a master regulator in QS pathways. 15
Targeting LsrR offers a promising strategy for disrupting QS and attenuating bacterial pathogenicity. Inhibitors designed to mimic AI-2 or disrupt LsrR binding to DNA can effectively interfere with its regulatory function. These small-molecule inhibitors either stabilize the inactive form of LsrR or prevent its interaction with the lsr operon, thereby hindering biofilm formation and reducing virulence. The specificity of these inhibitors makes LsrR an attractive target for developing anti-QS therapies aimed at combating biofilm-associated infections. 16 In this study, a structure-based virtual screening (SBVS) protocol was used to screen plant bioactive compounds based on the predicted active site of the target protein, followed by binding free energy calculations. The compounds selected from the SBVS were subjected to induced-fit docking to examine the dynamic nature of the protein–ligand interaction through flexibility. Molecular dynamics (MD) simulation is used to determine protein–ligand stability, which helped identify potent plant bioactive compounds that act as LsrR inhibitors by blocking its interaction with phospho-AI-2, thereby stabilizing LsrR-mediated repression, and preventing QS activation and biofilm formation in E. coli.
MATERIALS AND METHODS
Protein Preparation
The three-dimensional structure of the target protein (PDB ID: 4L4Y) with a resolution of 1.90 Å was downloaded from the Protein Data Bank and prepared using the Protein Preparation Wizard module in the Schrödinger Suite. The bond orders were assigned, hydrogen atoms were added at the initial stage, and crystal water molecules were removed. Furthermore, the minimization process was performed after optimization until the average root mean square deviation (RMSD) of the nonhydrogen atoms reached 0.3 Å. 17
Active Site Prediction
The binding site of the LsrR target protein was predicted using the Sitemap module in the Schrödinger software suite. Sitemap identifies energetically favorable regions by analyzing the interaction energies. Initially, potential binding sites were generated by placing site points on a grid. For this analysis, the number of site points per site was set to 15, and potential sites were evaluated. The calculations employed a more stringent hydrophobicity definition and utilized the Optimized Potential for Liquid Simulations (OPLS) force field. 18
Ligand Preparation
The LigPrep module was employed to prepare the ligand collected from the literature. The original state and chirality of the ligands were preserved by applying the optimized potential for the liquid simulation OPLS-3 force field. 19
Structure-Based Virtual Screening
To perform SBVS, the Grid-Based Ligand Docking with Energetics (Glide) module was executed. To soften the potential of the target region, we maintained the van der Waals radii of the receptor atoms at 1.00 Å with a partial charge cutoff of 0.25 to soften the potential for the target region. A grid was generated at the centroid of the binding site, and the prepared ligands were docked into the binding site of the target protein using high-throughput virtual screening (HTVS), standard precision (SP), and extra precision (XP). Glide generates the conformation, and all the conformations are allowed into a series of filters, and the final best-docked pose was selected using the scoring function parameters. 20
Molecular Mechanics–Generalized Born Surface Area Calculations
The Prime suite was employed to compute the binding free energy calculations for the docked protein–ligand complex, which was calculated using the equation given below:
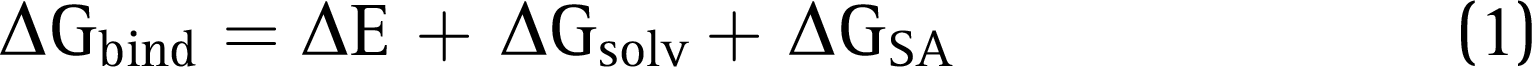
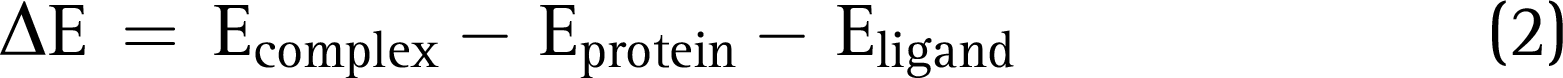
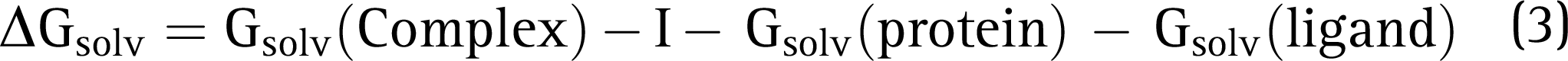
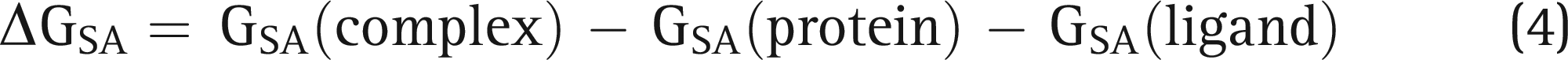
Induced-Fit Docking
Initially, each ligand was docked using a softened potential, where the van der Waals radii were scaled to allow for flexibility. Subsequently, side-chain predictions were performed for residues located within a specified distance of each ligand pose. Subsequently, minimization was performed on the same set of residues and ligands within each protein–ligand complex pose. At this stage, receptor structures adopt conformations that are optimized to fit the ligand. Finally, the ligand was subjected to a rigorous docking procedure using Glide XP to target the induced-fit receptor structures. During the initial docking phase, the van der Waals radii were scaled by a factor of 0.5 for both the receptor and ligand to enhance flexibility. The Prime refinement stage was applied to the side chains of the residues within 5 Å of the ligand, ensuring precise modeling of the interactions. Up to 20 poses were retained for each ligand during the initial docking, and these poses were subsequently redocked using Glide XP to enhance accuracy. 22 In this research, phospho-AI-2 was redocked into the 4L4Y structure to reproduce the cognate binding pose and used this validated site to define the docking grid for subsequent screening (Supplementary Fig. S1). This adjustment strengthens the robustness of our workflow and ensures that our results are directly grounded in experimental structural data.
MD Simulations
The stability of the protein–ligand complexes was evaluated using Molecular dynamics (MD) simulations carried out with the Desmond module (Schrödinger LLC). Each complex was prepared in an orthorhombic periodic boundary box and solvated with TIP3P water molecules, ensuring at least a 10 Å buffer between the protein surface and the box edges to avoid interactions with periodic images. The system was neutralized and adjusted to a physiological ionic strength by adding 0.15 M NaCl. Ligand parameters were generated using the OPLS_2005 force field, which was also applied to the protein. The system underwent initial energy minimization using the steepest descent algorithm to remove unfavorable contacts. Subsequently, equilibration was performed in multiple stages under the NPT ensemble, where the temperature was maintained at 300 K with the Nosé–Hoover chain thermostat and the pressure was controlled at 1 atm using the Martyna–Tobias–Klein barostat. A time step of 2 fs was used, and all covalent bonds involving hydrogen atoms were constrained with the SHAKE algorithm to allow stable integration of the equations of motion. The equilibrated system was then subjected to produce MD runs, during which the trajectory was collected for downstream analyses of protein–ligand stability 23
RESULTS AND DISCUSSION
Active Site Prediction
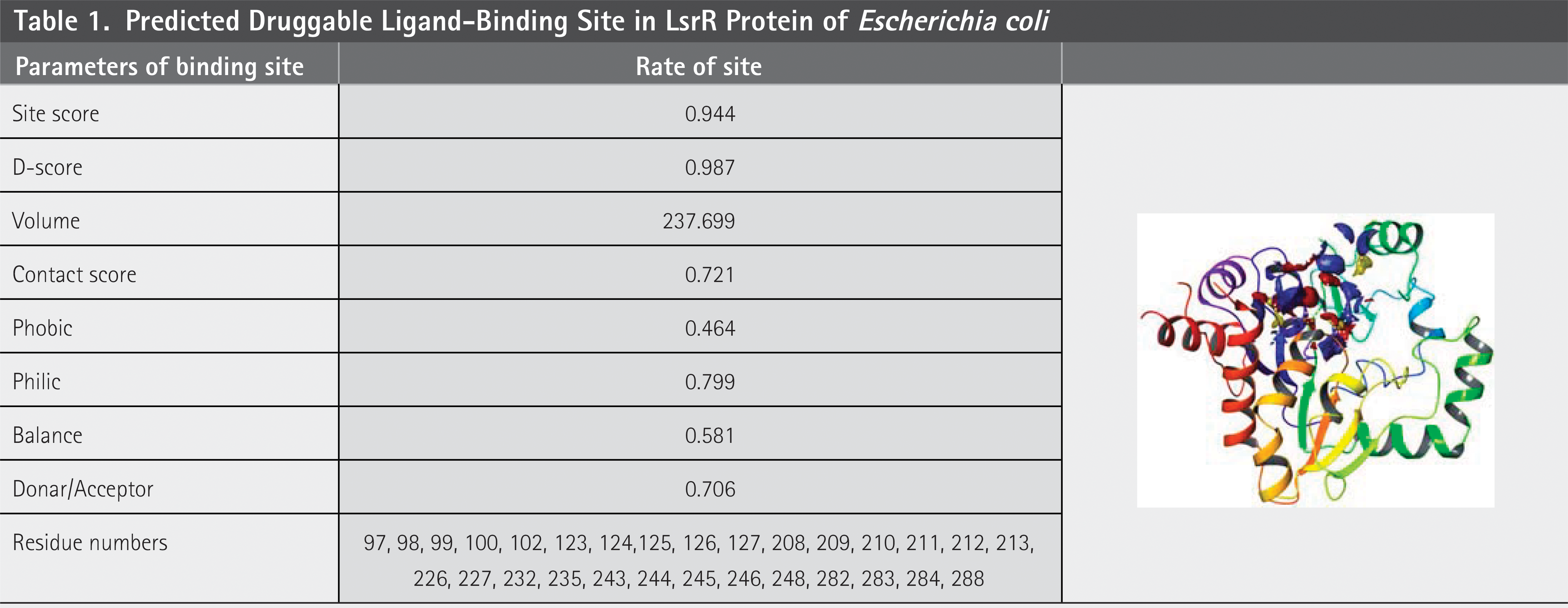
The Sitemap module was used to predict the active sites of the target proteins. The quality of the predicted active sites was assessed using several parameters ( Table 1 ). Specifically, the site score and druggability score (D-score) were 0.994 and 0.987, respectively, which are close to 1.00, indicating that the target protein possesses a high-quality binding site suitable for ligand interaction. The active site residues are listed in Table 1 .
Predicted Druggable Ligand-Binding Site in LsrR Protein of Escherichia coli
Electrostatic Potential Surface of Protein
The electrostatic potential surface of the binding site was predicted using molecular mechanics, which revealed electropositive and electronegative potential regions of the protein. The predicted electrostatic potentials of the target proteins are shown in Figure 2 . Here, the blue color depicts the electropositive region, and the red color indicates the electronegative region, which plays a crucial role in protein–ligand interactions.
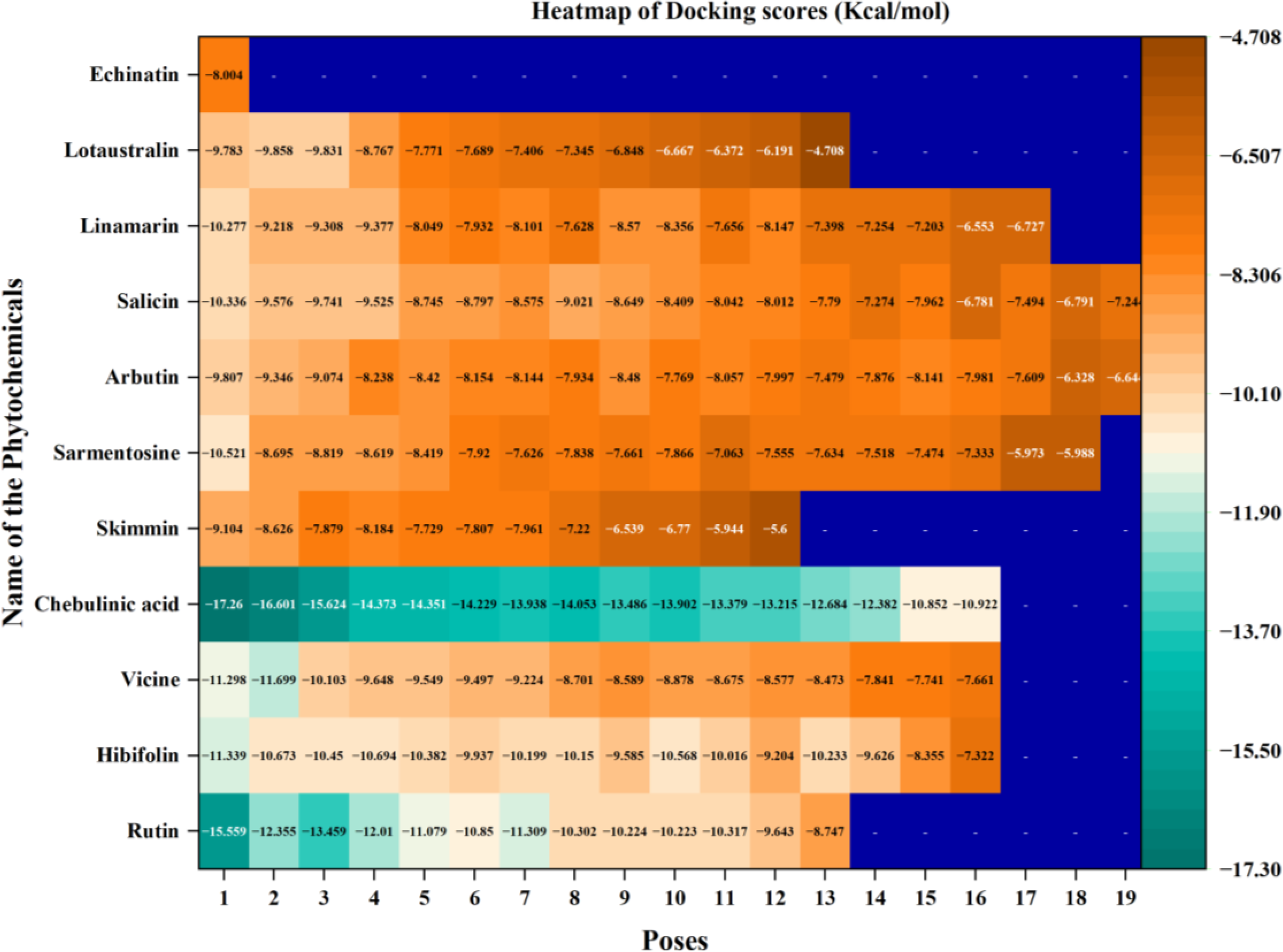
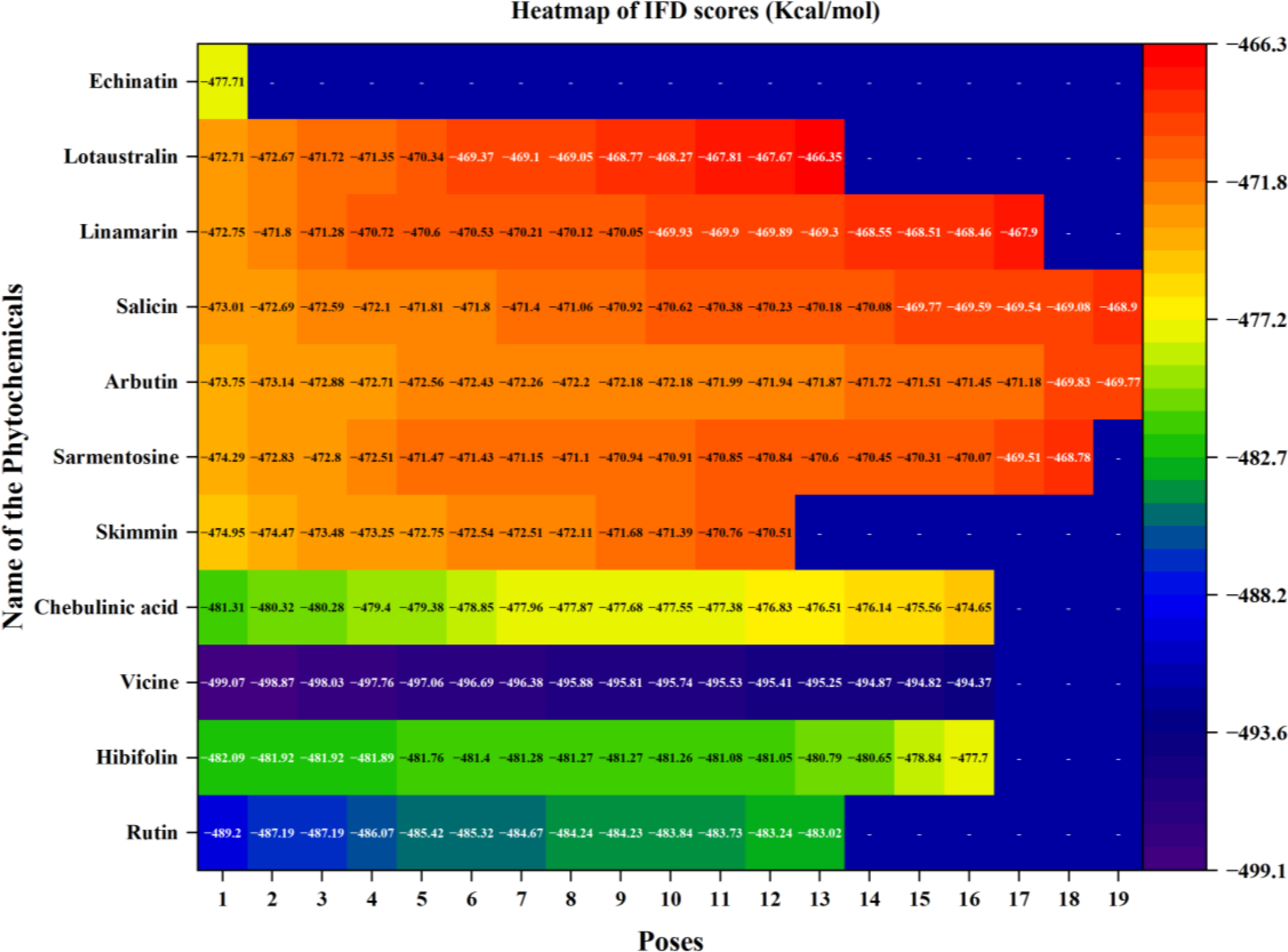
The heatmap represents the docking scores of phytochemicals at various binding poses in the quorum sensing LsrR protein of E. coli.
Structure-Based Virtual Screening
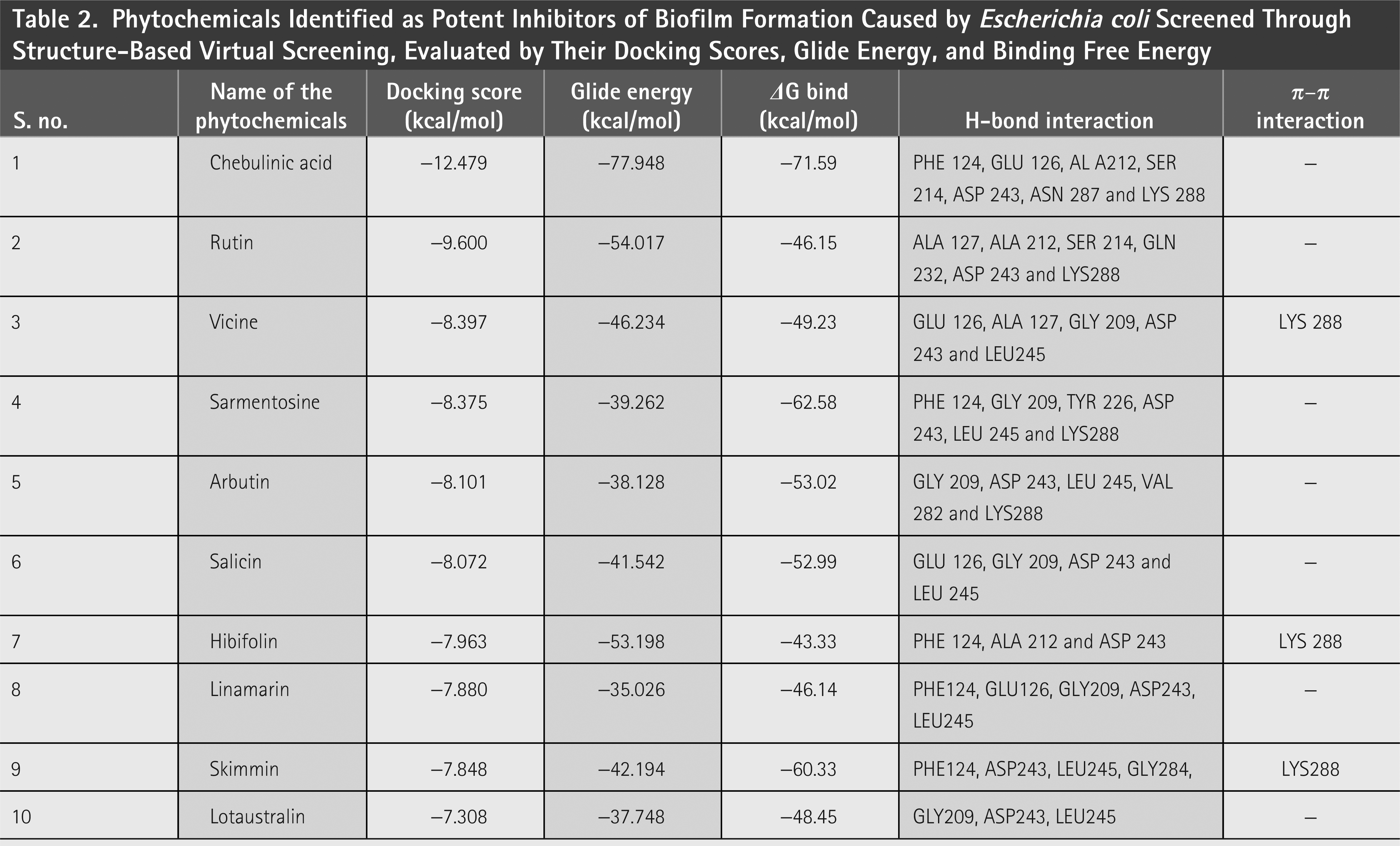
SBVS was performed to identify potential plant bioactive compounds from the constructed plant compound library obtained from the literature. Initially, HTVS was performed, which helped to screen the compounds quickly, and compounds with good docking scores were subjected to SP docking. Furthermore, compounds with better docking scores were subjected to XP docking. The best screened compounds are tabulated in Table 2 .
Phytochemicals Identified as Potent Inhibitors of Biofilm Formation Caused by Escherichia coli Screened Through Structure-Based Virtual Screening, Evaluated by Their Docking Scores, Glide Energy, and Binding Free Energy
Binding Mode Analysis
The reported (echinatin) and screened compounds were docked into the binding site of the protein, and it was found that the plant bioactive compounds obtained from the screening had better docking scores than echinatin. Echinatin has been previously identified as having potential for tackling AMR bacteria by preventing biofilm formation in E. coli and Staphylococcus aureus without affecting the bacteria themselves. It serves as a QS inhibitor, disrupting bacterial communication that facilitates biofilm formation and pathogenicity. 24 Consequently, echinatin was used to compare the potential of selected phytochemicals in the present research. The protein–ligand interactions also clearly indicated that the screened compounds had better interactions than the reported compounds. Furthermore, the 10 screened compounds were subjected to an induced-fit docking (IFD) protocol.
Binding Free Energy Calculation
The energy associated with the protein–ligand interactions was determined through binding free energy calculations. Table 3 presents the docking outcomes and the corresponding binding free energy values. A significant correlation was observed between the binding free energy (ΔG) values and docking scores of the receptor–ligand complexes.
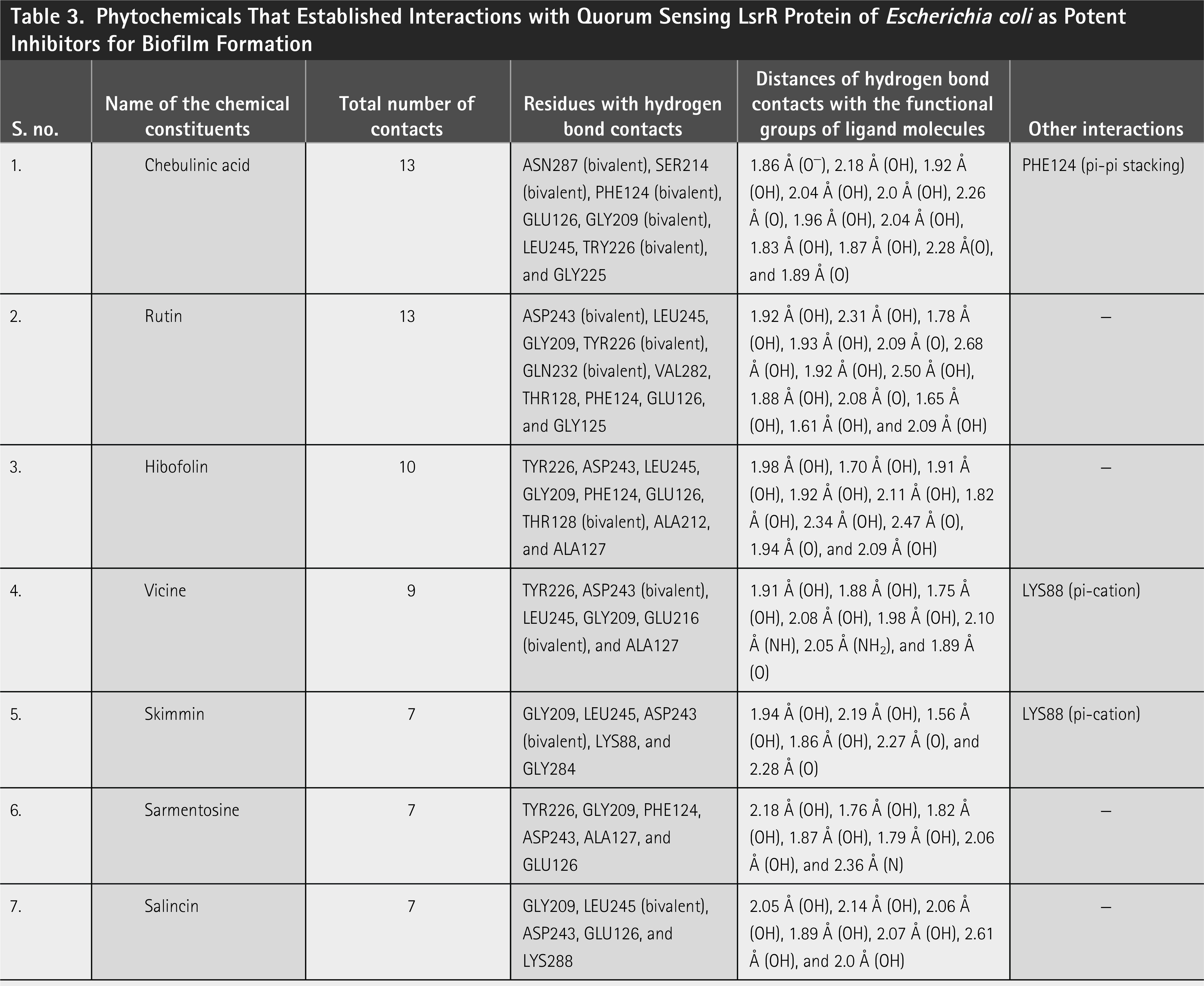
Phytochemicals That Established Interactions with Quorum Sensing LsrR Protein of Escherichia coli as Potent Inhibitors for Biofilm Formation
Induced-Fit Docking
IFD provides a good method to obtain changes at the interaction interface when the protein recognizes the ligand. The results of the IFD are presented in Supplementary Figure S1 and Figure 2 .
Chebulinic acid
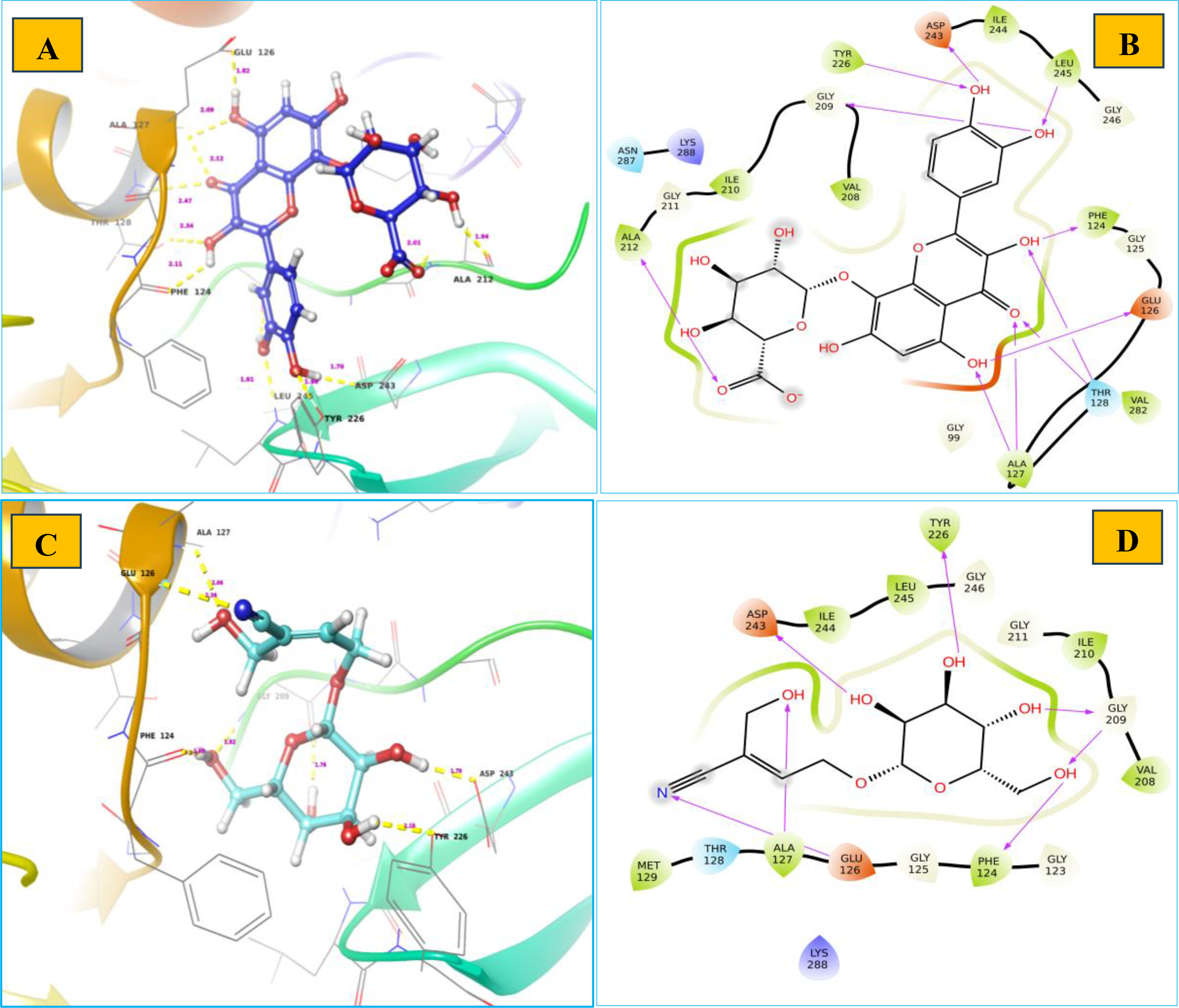
Chebulinic acid exhibited strong binding in 18 out of 20 poses, with notable docking scores ranging from −17.260 to −5.988 kcal/mol ( Fig. 2 ). Notably, it had a docking score of −17.260 kcal/mol with an IFD score of −481.31 kcal/mol ( Fig. 3 ) in pose 1, including 12 hydrogen bond contacts and a π–π stacking contact. The residues that interacted with this molecule were ASN287 (bivalent), SER214 (two interactions), PHE124 (two interactions), GLU126, GLY209 (two interactions), LEU245, TRY226 (two interactions), and GLY225 ( Table 3 ). The distances of hydrogen bond contacts between these residues and the functional groups of chebulinic acid were 1.86 Å (O−), 2.18 Å (OH), 1.92 Å (OH), 2.04 Å (OH), 2.0 Å (OH), 2.26 Å (O), 1.96 Å (OH), 2.04 Å (OH), 1.83 Å (OH), 1.87 Å (OH), 2.28 Å(O), and 1.89 Å (O), respectively ( Fig. 4A , B).
The heatmap represents the induced-fit docking scores of phytochemicals at various binding poses in the quorum sensing LsrR protein of E. coli.

Binding affinities of phytochemicals with quorum sensing LsrR protein of E. coli:
Chebulinic acid is an ellagitannin that is widely found in various plants, particularly in Terminalia chebula fruits. 25 It is well known for its various pharmacological properties, including anticancer, antidiabetic, antiatherogenic, antiulcer, antifibrotic, hepatoprotective, antioxidant, anti-inflammatory, antiepithelial, neurodegenerative disease protective, antituberculotic, and antibacterial activities. 25 In a recent study, Ou et al. 26 reported that it effectively inhibited Helicobacter pylori with a minimum inhibitory concentration (MIC) of 32 µg/mL at a dose of 5 µg/mL, implying strong antibacterial properties. Ou et al. 26 further confirmed the antibacterial effects of this chemical against H. pylori using a drug design approach involving in silico molecular docking. In their in silico study, it had a strong binding affinity and a docking score of −9.7 kcal/mol for the virulence factor associated with the pathogenicity of H. pylori, especially cytotoxin-associated gene A. This outcome further confirms that chebulinic acid exerts potent antibacterial properties by targeting bacterial virulence factors. 26 In an in silico study, it was reported to have antituberculotic properties, as it established strong binding affinities with the DNA gyrase of Mycobacterium tuberculosis. In addition, it had a notable docking score of −14.63 kcal/mol. 27 According to Vu et al. 28 it has antibacterial properties that inhibit the growth of Acidovorax avenae. Cattleyae, Agrobacterium tumefaciens, Burkholderia glumae, Pectobacterium carotovorum carotovorum, P. chrysanthemi, Pseudomonas syringae pv. actinidia, P. solanacearum, and Xanthomonas arboricola pv. Pruni. The MICs for these strains were 250, 250, 250, 250, 250, 104.2, 250, 52.1, and 52.1 µg/mL at concentrations ranging from 7.8 to 250 μg/mL.
Rutin
Rutin exhibited strong binding in 13 of the 20 poses, with docking scores ranging from −15.559 to −8.747 kcal/mol ( Table 3 and Fig. 2 ). Notably, it had a docking score of −15.559 with an IFD score of −489.20 kcal/mol ( Fig. 3 ). Pose 1 showed 13 hydrogen bond interactions with LsrR. The residues that interacted with rutin were ASP243 (two interactions), LEU245, GLY209, TYR226 (two interactions), GLN232 (two interactions), VAL282, THR128, PHE124, GLU126, and GLY125 ( Table 3 ). The distances of hydrogen bond contacts between these residues and the functional groups of rutin were 1.92 Å (OH), 2.31 Å (OH), 1.78 Å (OH), 1.93 Å (OH), 2.09 Å (O), 2.68 Å (OH), 1.92 Å (OH), 2.50 Å (OH), 1.88 Å (OH), 2.08 Å (O), 1.65 Å (OH), 1.61 Å (OH), and 2.09 Å (OH) ( Fig. 4C , D). Rutin is a flavonoid that is found in several fruits, vegetables, and other botanicals. However, the highest rutin volume was recorded in Fagopyrum esculentum (grains) and Amaranthus spp. (leaves), Asparagus officinalis (rhizome) and Citrus spp. (fruit), Ruta graveolens, and Morus alba. It is primarily known for its antioxidant potential because it belongs to the flavonoid class. 29 Nonetheless, it has also been documented to exhibit antibacterial properties against S. aureus (MIC, 512 µg/mL), S. epidermidis (MIC, 512 µg/mL), Enterococcus faecalis (MIC, 256 µg/mL), Bacillus subtilis (MIC, 256 µg/mL), E. coli (MIC, 512 µg/mL), Klebsiella pneumoniae (MIC, 256 µg/mL), Proteus mirabilis (MIC, 256 µg/mL), Enterobacter cloacae (MIC, 128 µg/mL), Pseudomonas aeruginosa (MIC, 1,024 µg/mL), and Acinetobacter baumannii (MIC, 1,024 µg/mL). 30 With MIC values of 80 and 400 µg/mL, it effectively inhibited the growth of Bacillus cereus and Salmonella enteritidis. 31 Although rutin has demonstrated inhibitory potential against various bacterial pathogens in vitro, its specific mechanisms of action have not yet been thoroughly investigated. However, an in silico study reported that rutin is effective in inhibiting S. aureus as it establishes a strong binding affinity with the key enzymes of S. aureus (transferase, ligase/RNA, isomerase, and oxidoreductase) responsible for pathogenicity, replication, and virulence factors. 32 The docking scores of rutin against these proteins ranged from −8.48 to −5.57 kcal/mol.
Vicine
Vicine exhibited strong binding in 16 of the 20 poses, with notable docking scores ranging from −11.699 to −7.661 kcal/mol ( Fig. 2 ). In this complex, pose 1 of vicine was selected for further analysis because of its substantial number of interactions. Despite pose 1 having a lower docking score of −11.298 kcal/mol and an IFD score of −499.07 kcal/mol ( Table 3 and Fig. 3 ) compared with position 2, it had good binding affinities with eight hydrogen bond interactions and one π–cation interaction. The residues that interacted with this molecule were TYR226, ASP243 (two interactions), LEU245, GLY209, GLU216 (two interactions), and ALA127 ( Table 3 ). The distances of hydrogen bond contacts between these residues and the hydroxyl group (OH), amine group (NH), and oxygen groups of vicine are 1.91 Å (OH), 1.88 Å (OH), 1.75 Å (OH), 2.08 Å (OH), 1.98 Å (OH), 2.10 Å (NH), 2.05 Å (NH2), and 1.89 Å (O), respectively ( Fig. 4E , F). Vicine is categorized as an alkaloid glycoside because it contains both an alkaloid and sugar molecule. It was first isolated from the seeds of Vicia sativa. The pyrimidine ring is an essential chemical constituent of the therapeutic agents. Vicine is a natural metabolite containing a pyrimidine ring isolated from V. sativa and V. faba. In 1984, Bjerg 33 investigated the antifungal properties of vicine and convicine against the fungi Ascochyta fabae, Botrytis cinerea, and Pyrenophora graminea and observed inhibited growth in all three species. Pavlík et al. 34 reported that C. herbarum, B. cinerea, and P. graminea were significantly inhibited by vicine at a lower concentration of 25 µg/mL. In addition, they noted some hyphae with interrupted growth in F. solani, F. culmorum, and A. alternata, respectively.
Hibifolin
Hibifolin exhibited strong binding in 16 of the 20 poses, with notable docking scores ranging from −11.339 to −7.322 kcal/mol ( Fig. 2 ). Notably, it had a docking score of −11.339 kcal/mol and an IFD score of −482.09 kcal/mol in pose 1 ( Fig. 3 ). It forms hydrogen bonds with TYR226, ASP243, LEU245, GLY209, PHE124, GLU126, THR128, ALA212, and ALA127 ( Table 3 and Fig. 5A ). The respective distances of these contacts are 1.98, 1.70, 1.91, 1.92, 2.11, 1.82, 2.34, 2.47, 1.94, and 2.09 Å. Most interactions occurred with the hydroxyl groups of hibifolin ( Fig. 5B ). According to Song et al., 35 hibifolin can inhibit SrtA activity, with an IC50 of 31.20 µg/mL. Furthermore, hibifolin was found to have the capacity to adhere to bacteria in host cells and notably reduce biofilm formation in treated USA300 cells. According to the results of molecular docking and fluorescence quenching assays, hibifolin can directly target SrtA. 35 This interaction was further corroborated by the observation that the inhibitory effects of hibifolin on mutant SrtA were significantly diminished after mutation of the binding sites (TRP-194, ALA-104, THR-180, ARG-197, and ASN-114). In vivo, combined administration of hibifolin and cefotaxime conferred significantly greater protection against USA300-induced pneumonia in mice than cefotaxime alone, whereas no cytotoxic effects were observed with hibifolin treatment. Song et al. 35 concluded that hibifolin attenuates the pathogenicity of methicillin-resistant S. aureus by directly targeting SrtA. Hence, they proposed that it could be utilized in the future as an adjuvant therapy for S. aureus infections.
Binding affinities of phytochemicals with quorum sensing LsrR protein of E. coli:
Sarmentosine
Sarmentosine exhibited strong binding in 18 of 20 poses, with notable docking scores ranging from −10.521 to −5.988 kcal/mol. Notably, it had a docking score of −10.521 and an IFD score of −474.29 kcal/mol ( Figs. 2 and 3 ). Pose 1 had seven hydrogen bond interactions with LsrR. The residues that interacted with sarmentosine are TYR226, GLY209, PHE124, ASP243, ALA127, and GLU126 ( Table 3 and Fig. 5A ). The distances of hydrogen bond contacts between these residues and the functional groups of sarmentine were 2.18, 1.76, 1.82, 1.87, 1.79, 2.06, and 2.36 Å, respectively. Except for GLU126, all residues were in contact with the hydroxyl groups of sarmentosine ( Fig. 5B ).
MD Simulations
Stability of chebulinic acid–QS LsrR of E. coli
In this complex, C-α had a low RMSD value of <1.7 Å for 100 ns. Overlapping ligand and protein RMSDs revealed that the complex exhibited strong binding stability from 0 to 100 ns ( Fig. 6A ). GLU 69, ALA 96, and SER 317 were the primary residues that exhibited fluctuations under thermostatic conditions, with displacements of 4.923, 4.29, and 4.035 Å, respectively ( Fig. 7A ). Notably, ALA 96 maintained its interaction with chebulinic acid, indicating its binding stability. ASP 243 had the highest interaction fraction range of 2.05 Å with water-assisted hydrogen-bond contacts, followed by GLU 126 and GLN 232 ( Fig. 8A ). Notably, ASP 243 exhibited stronger hydrogen bonding interactions with the hydroxyl groups of chebulinic acid, with binding strengths of 96% and 87%, respectively ( Fig. 8B ). This complex exhibited good binding stability with 18 interactions and an average of 12 contacts ( Fig. 8C ).
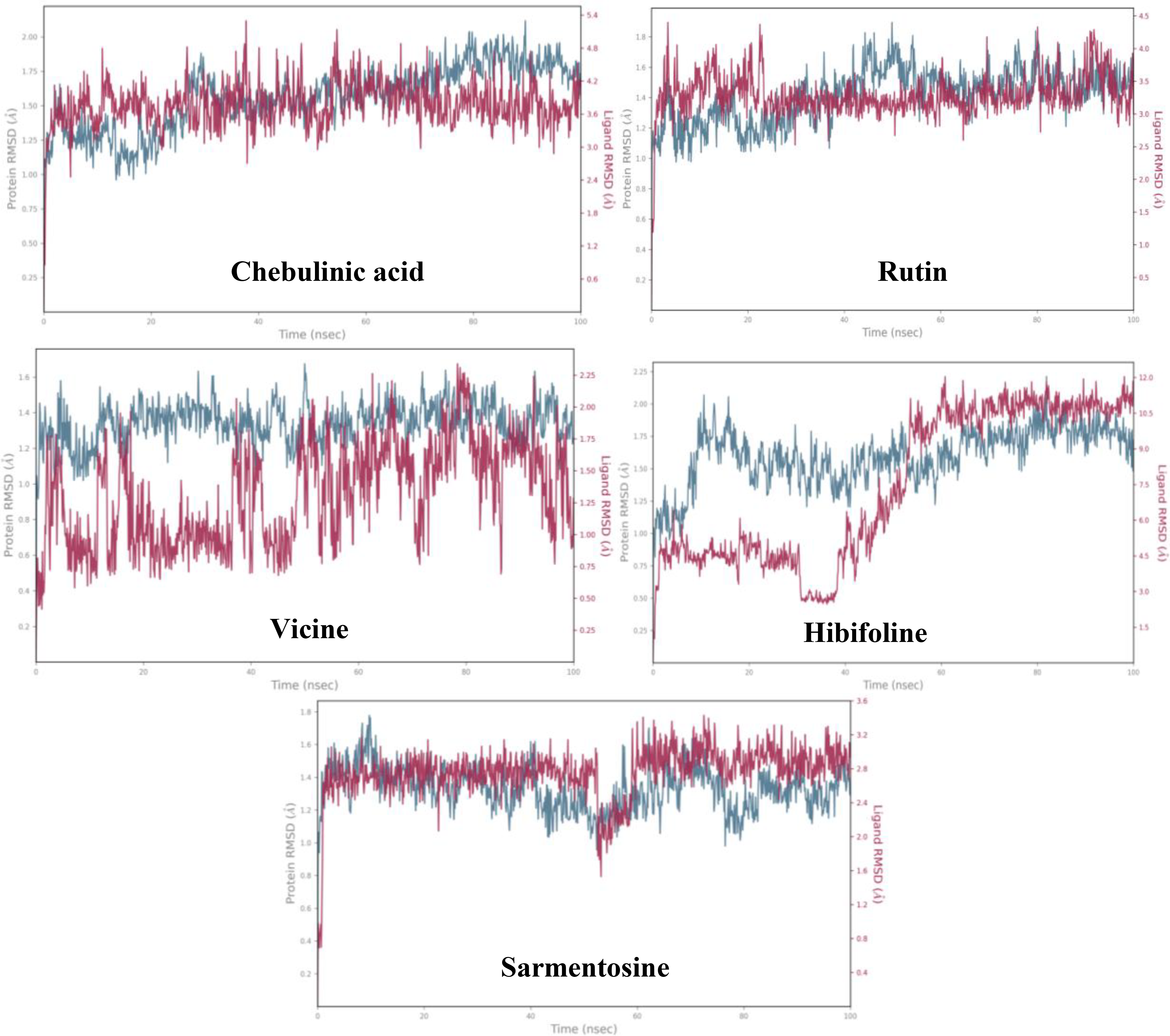
The plots represent the root mean square deviation ranges of the phytochemicals that hit the quorum sensing LsrR protein of E. coli for a simulation periods of 100 ns.
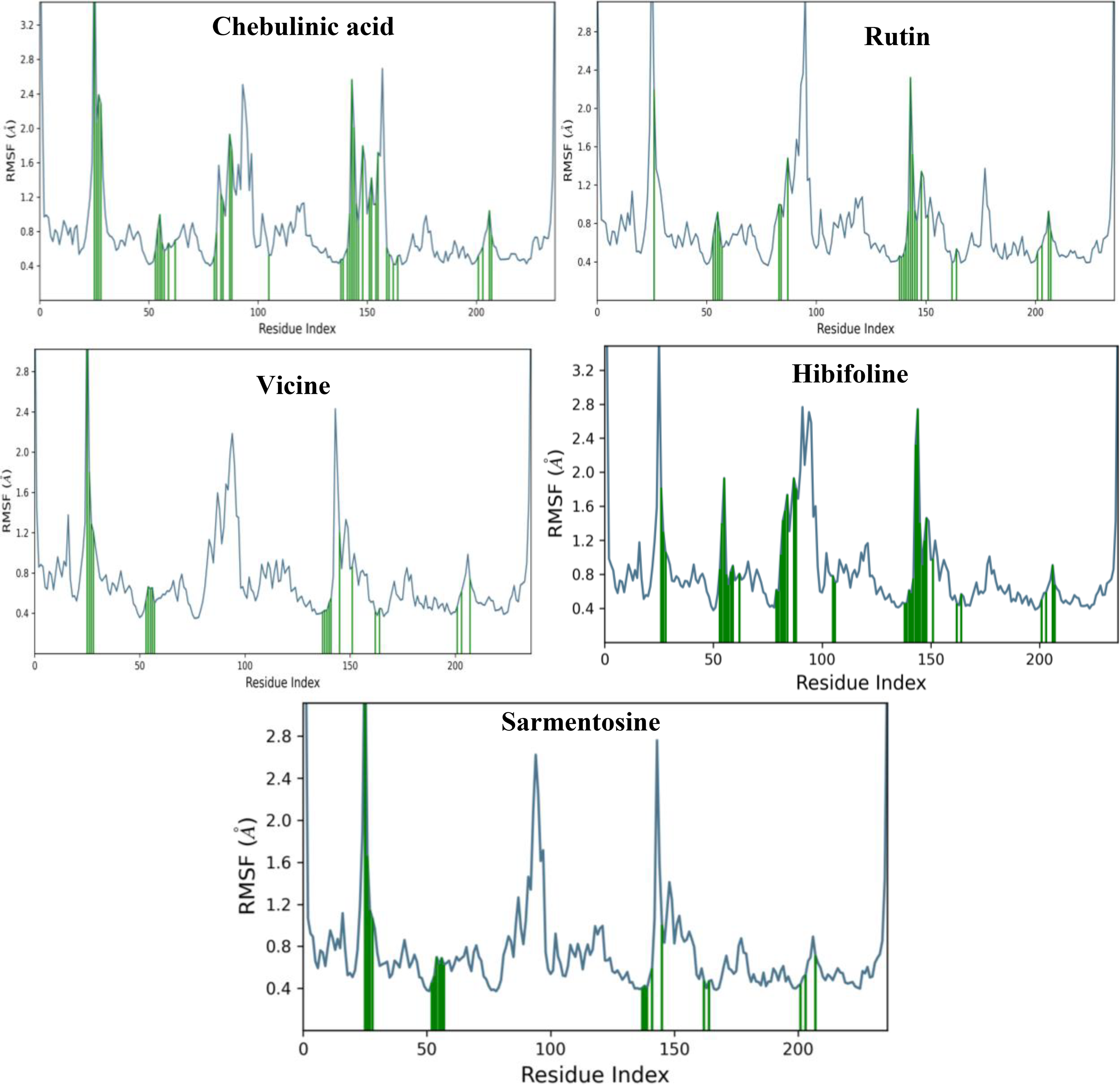
Plots represent the most influenced residues of top hits-quorum sensing LsrR protein of E. coli complexes during the simulation periods.
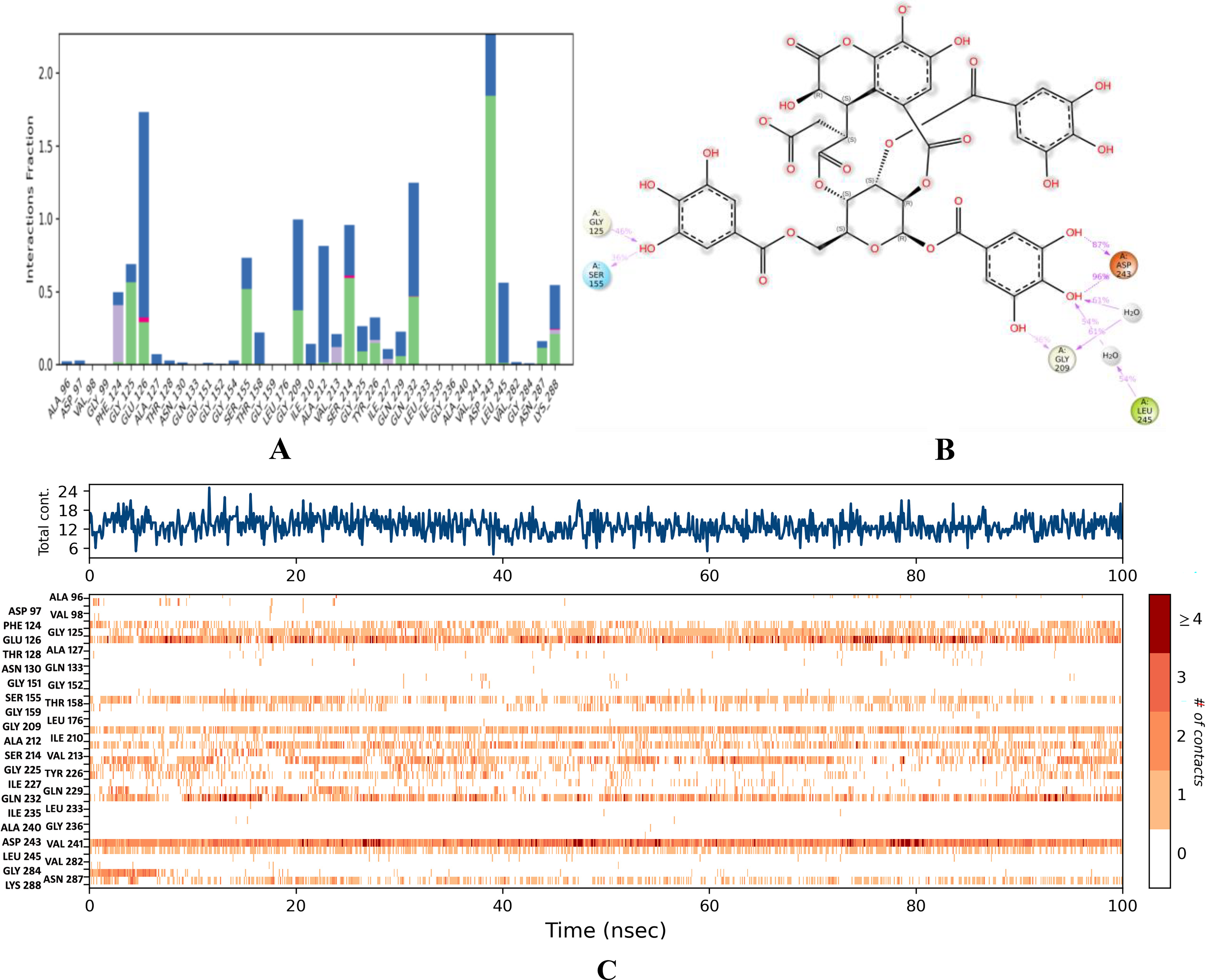
Binding stability of chebulinic acid.
Stability of rutin–QS LsrR of E. coli
Simulation analysis revealed that the rutin–QS LsrR complex had good binding stability, with an RMSD value of <1.6 Å ( Fig. 6B ). The initial RMSD range of C-α for this complex from 0 to 47 ns was <1.4 Å. Subsequently, a minor deviation was detected at 47 ns, which reached a RMSD of 1.6 and persisted until 53 ns. Nevertheless, it reverted to the initial RMSD range of 1.4 Å and persisted until 100 ns. Notably, the overlapping protein and ligand RMSDs suggested enhanced stability in the binding affinities of rutin–QS LsrR ( Fig. 6B ). In this complex, residues SER 317, ALA 96, GLU 69, and ALA 166 were identified as highly mobile regions in the QS LsrR protein of E. coli, exhibiting fluctuations of up to 4.381, 3.925, 3.859, and 3.222 Å, respectively ( Fig. 7B ). ASP 243 had the highest interaction range of 2.6 Å, solely through hydrogen bond contacts ( Fig. 9A ). Similarly, GLY 209 exhibited only hydrogen-bond interactions, with a range of 1.1 Å. In the post-MD interaction plot, ASP 243 (99, 99, and 72%), 124 (98%), 209 (96%), and 212 (87%) exhibited strong binding affinity with rutin ( Fig. 9B ). The complex exhibited considerable stability with an average of 13 contacts ( Fig. 9C ). Notably, the residues that consistently maintained contact with rutin throughout the simulation period are indicated by the dark red lines in the interaction timeline plot.
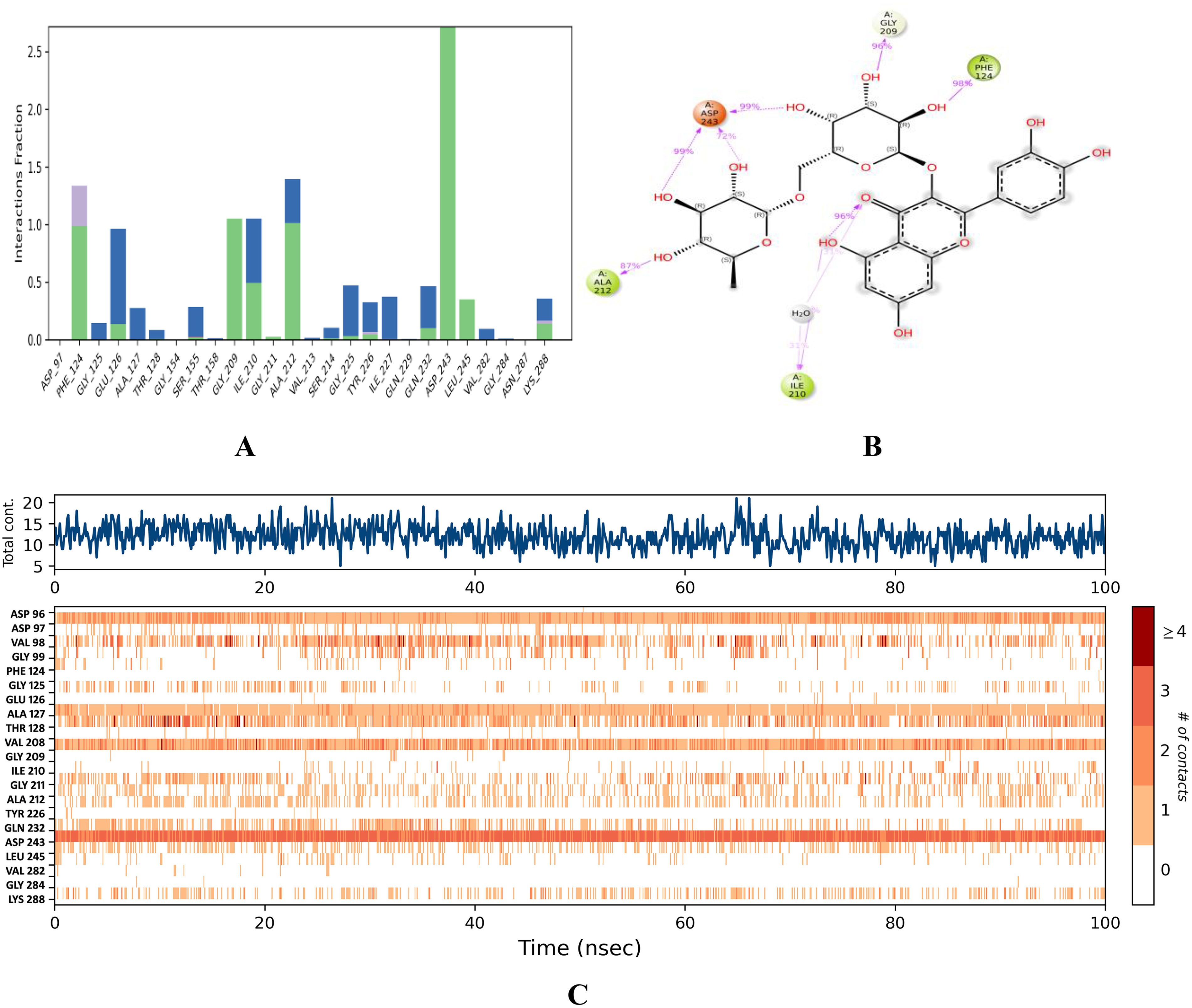
Binding stability of rutin.
Stability of vicine–QS LsrR of E. coli
The C-α of this complex consistently exhibited an RMSD peak of 1.4 Å ( Fig. 6C ), indicating stronger stability of the binding affinity. Although the ligand RMSD exhibited deviations from 0 to 50 ns, it subsequently aligned with the protein RMSD at 50 ns and maintained its stability until 100 ns, further corroborating the binding stability of this complex. Notably, thermostatic conditions had no effect on the binding affinity of this complex, which is a testament to its strong binding affinity. In this complex, residues GLU 69, ALA 96, and SER 317 exhibited the most significant fluctuations, with movements of 4.083, 3.773, and 3.659 Å, respectively ( Fig. 7C ). In this case, ALA 96 maintained contact with vicine under thermostatic and pressure conditions. ASP 243, PHE 124, GLU 126, and GLY 209 had notable interaction fraction values of 1.80 Å, 1.72 Å, and 1.58 Å, respectively ( Fig. 10A ). These residues established strong water-assisted hydrogen bond contacts with strengths of 97, 91, 96, and 93%, respectively ( Fig. 10B ). This complex exhibited 12 interactions with an average of 10 contacts between 0 and 100 ns ( Fig. 10C ).
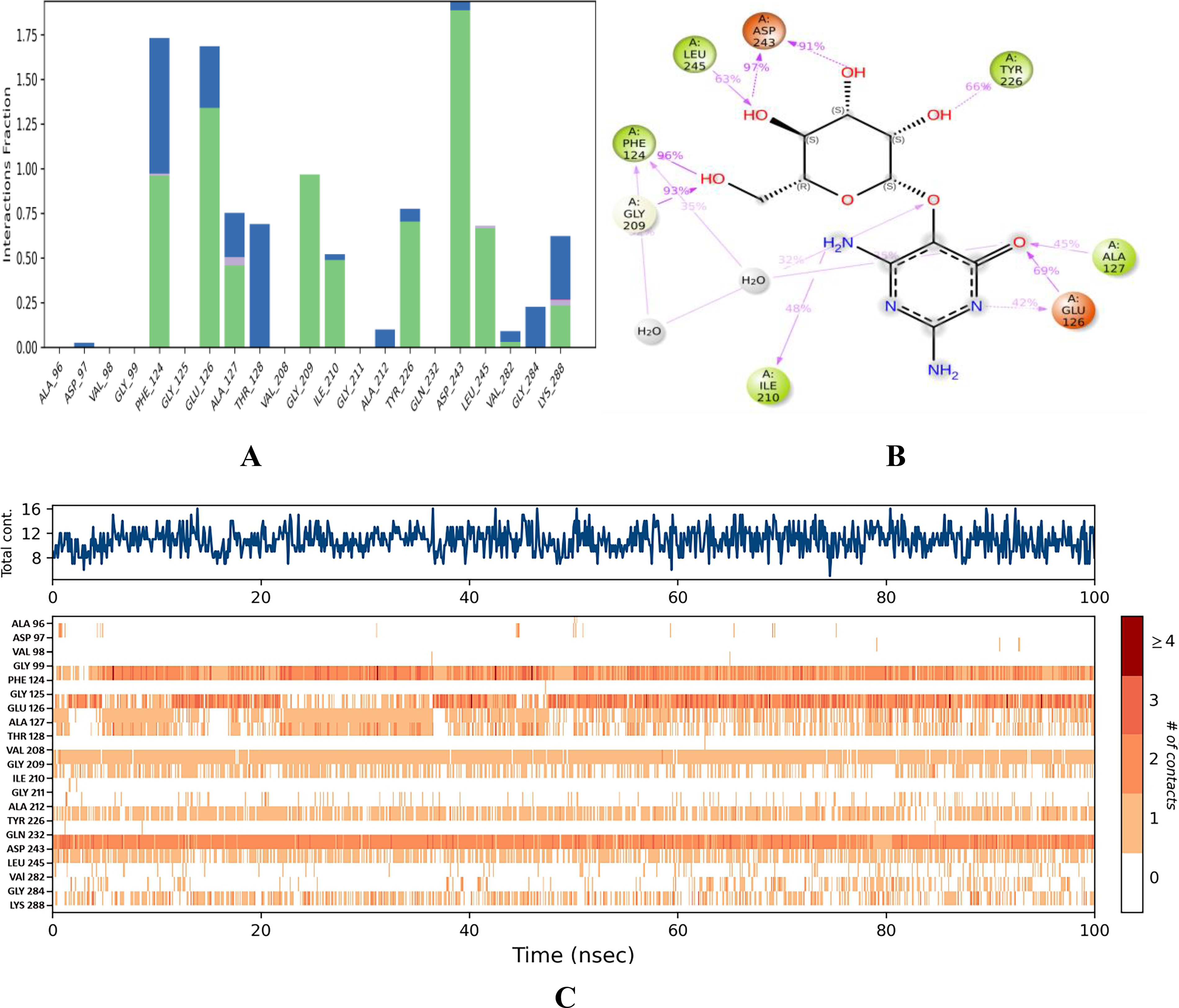
Binding stability of vicine.
Stability of hibifolin–QS LsrR of E. coli
In this complex, the initial RMSD was 1.25 Å from 0 to 7 ns. Subsequently, a larger deviation occurred at 9 ns, reaching an RMSD range of 2.0 Å ( Fig. 6D ). Subsequently, the RMSD gradually decreased and reached 1.60 Å at 18 ns. This persisted for up to 70 ns, with minor deviations. Interestingly, parallel lines of ligand and protein RMSD ranges were observed at 57 ns, which persisted for up to 100 ns. The Root Mean Square Fluctuation (RMSF) plot of this complex indicated that GLU 69, SER 317, GLY 70, and ALA 96 had the greatest movement, with ranges of 5.479, 4.121, 3.844, and 3.609 Å, respectively ( Fig. 7D ). PHE 124 formed water- and hydrophobic-assisted hydrogen bond contacts with hibifolin, showing an interaction fraction range of 1.0 Å ( Fig. 11A ). The post-MD interaction plot for this complex showed that none of the QS LsrR residues had a stronger connection to hibifolin than to chebulinic acid, rutin, and vicine ( Fig. 11B ). Similarly, the number of contacts for this complex was determined to be 12 from 0 to 50 ns, whereas the maximum number of contacts decreased from 50 to 100 ns ( Fig. 11C ).
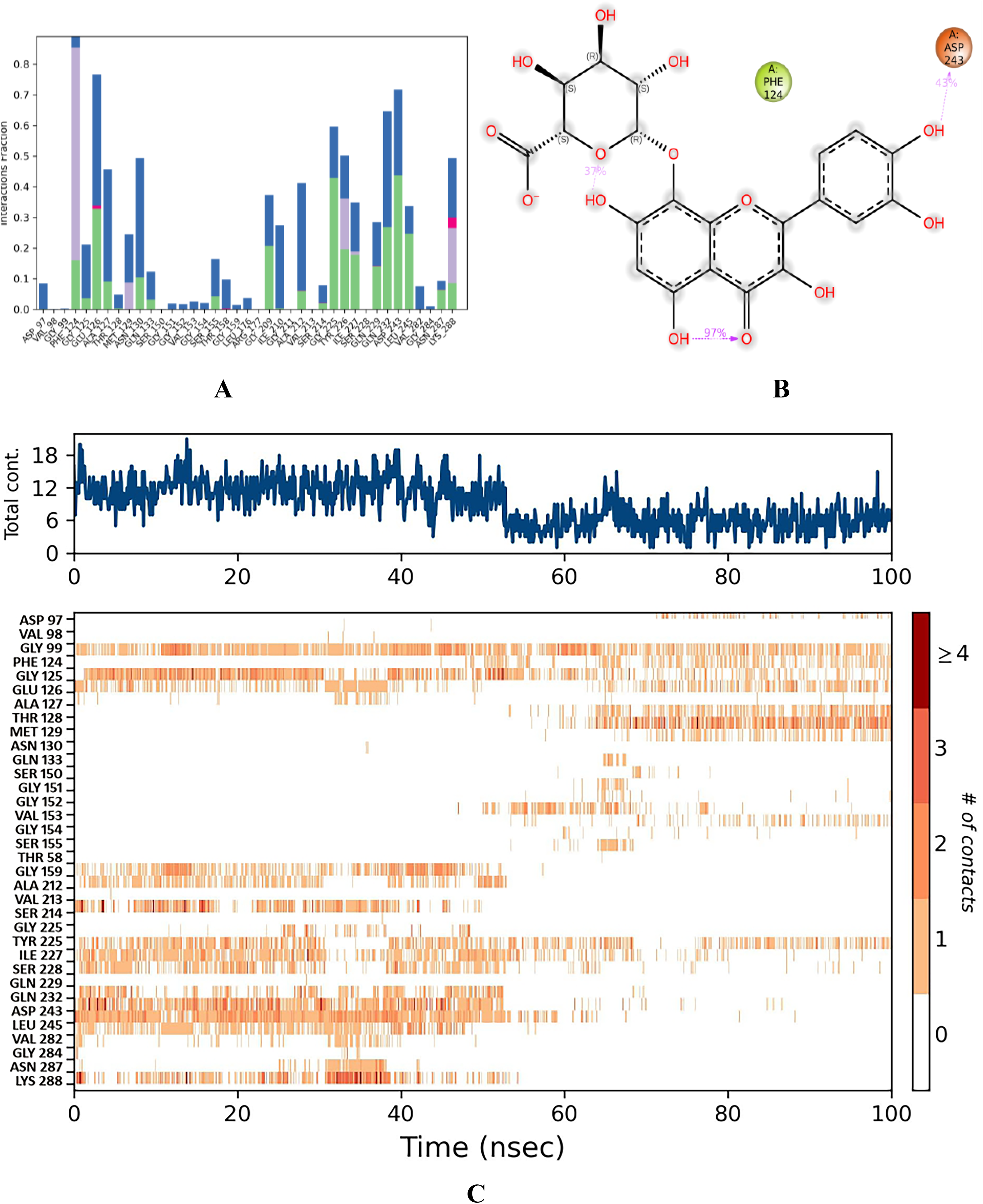
Binding stability of hibifolin.
Stability of sarmentosine–QS LsrR of E. coli
The C-α of this complex demonstrated remarkable stability, maintaining a consistent RMSD value of 1.4 Å throughout the 0–100 ns interval ( Fig. 6E ). However, a slight deviation that did not occur was observed at 50 ns. In addition, RMSD was within a stable range of <1.1 Å. Interestingly, the overlapping RMSD of the ligand and protein indicated stronger binding stability of this complex. The RMSF plot showed that GLU 69, GLY 70, ALA 96, and SER 317 were influenced by the thermostatic effects. Consequently, these residues exhibited the highest fluctuation ranges of 6.672, 4.115, 3.879, and 3.641 Å, respectively ( Fig. 7E ). Nonetheless, ALA 96 maintained its interaction with sarmentosin, even though it fluctuated by up to 3.879 Å. With interaction fraction ranges of 1.5, 1.0, 0.9, 0.84, and 0.8 Å, the amino acids ASP 243, GLY 209, PHE 124, TYR 226, and GLY 284 exhibited water-assisted hydrogen-bond interactions with sarmentosine ( Fig. 12A ). The residues that exhibited the strongest binding affinities for sarmentine were ASP 243 (89%), TYR 226 (76%), ALA 127 (68%), and GLY 284 (67%) ( Fig. 12B ). Throughout the simulation period, this complex exhibited an average of nine contacts with a maximum of 13 ( Fig. 12C ).
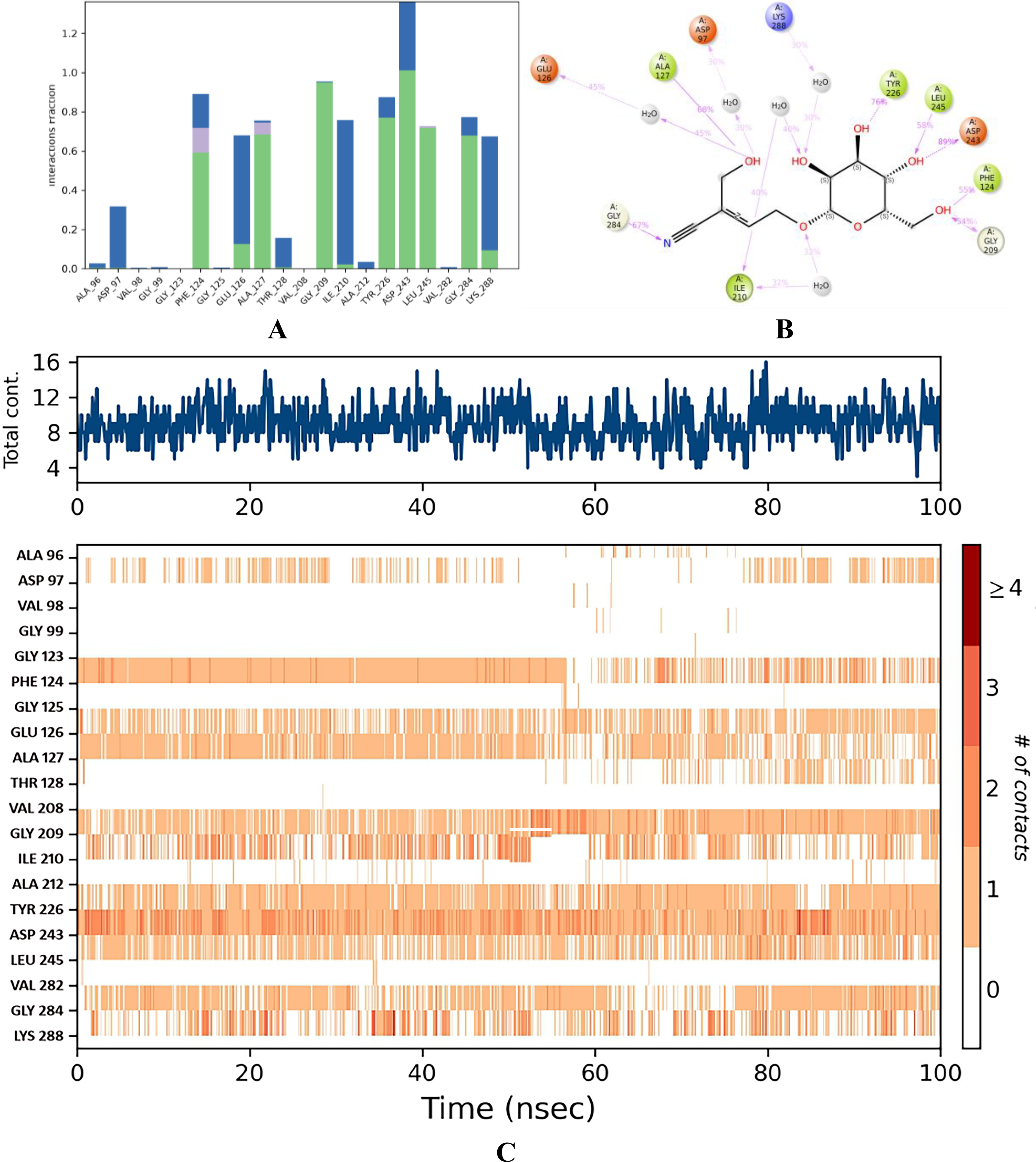
Binding stability of sarmentosine.
Molecular docking and MD simulations are essential techniques in drug discovery that facilitate the exploration of genetic responses, drug–receptor interactions, and biological processes.36,37 The use of these computational technologies not only reduces the costs associated with experimental procedures but also significantly enhances the efficiency of research and development processes.36,38 To support this conclusion, the current study employed molecular docking and MD simulations to elucidate the atomic-level interactions between QS LsrR protein of E. coli and hit phytochemicals. Eventually, these techniques have enabled the identification of viable therapeutic alternatives for combating biofilm formation in bacterial infections.
Inactive and active probability rate of organ toxicity and LD50 values of potent hits
To avoid adverse effects during and after treatment, the current study investigated the organ toxicity and toxicity endpoints of the phytochemicals identified in the present study. This study focused on the organ toxicity probabilities of phytochemicals with respect to hepatotoxicity, neurotoxicity, nephrotoxicity, respiratory toxicity, and cardiotoxicity. Similarly, toxicity endpoints were investigated, including carcinogenicity, immunotoxicity, mutagenicity, cytotoxicity, blood–brain barrier permeability, ecotoxicity, clinical toxicity, and nutritional toxicity. Chebulic acid, rutin, hibifolin, and sermentosin were found to have a low risk of inducing hepatotoxicity and neurotoxicity, with inactive probability rates of 0.87, 0.80, 0.75, and 0.73, as well as 0.89, 0.89, 0.90, and 0.70, respectively ( Table 4 ), indicating that these phytochemicals are extremely safe for the liver and neurons. Conversely, rutin and sermentopsine were identified as safer for both the cardiovascular and respiratory systems, with notably low inactivity probability rates of 0.98 and 0.73 ( Table 4 ), respectively. Regarding nephrotoxicity, respiratory toxicity, and cardiotoxicity, the remaining phytochemicals, except rutin and sermentine, demonstrated a modest probability of being either active or inactive. This indicated that phytochemicals may or may not elicit harmful effects. The assessment of toxicity endpoints revealed that chebulic acid and rutin exhibited immunotoxicity probability rates of 0.91 and 0.98 ( Table 4 ), respectively. This finding indicated a substantial likelihood of immunotoxicity induced by these compounds. Interestingly, vicine, hibifolin, and sarmentosine were found to be safer drug candidates for microbial infections, as they showed moderate to high inactive probability rates for organ toxicity and toxicity endpoints, indicating a lower risk of the explored toxicities. In particular, the Lethal Dose 50% (LD50) value of sarmentosine is 23,000 mg/kg, indicating that it has a lower fatal risk to humans compared with all phytochemicals analyzed in the present investigation ( Table 4 ). Toxicity assesses the harmful effects of chemicals on humans, animals, and the environment, with the toxicity endpoint serving as a specific measure for evaluating multiple toxicities, including those of carcinogenicity, cytotoxicity, and genotoxicity. 40 It is also possible to measure the deadly dose of substances for humans, such as the LD50 (lethal dose for 50% of subjects tested). In silico approaches are used to enhance in vitro and in vivo toxicity assessments, reduce the need for animal testing, lower the cost-effectiveness and time required for toxicity evaluations, and improve toxicity prediction and safety analysis. Furthermore, in silico assessments offer the distinct benefit of predicting the toxicity of a compound before it is synthesized. 41
Organ Toxicity and Toxicity Endpoints of Phytochemicals Identified for the Inhibition of Escherichia coli Biofilm Formation Targeting Quorum Sensing LsrR Protein (Predicted in ProTox 3.0 According to Banerjee et al. 39 )
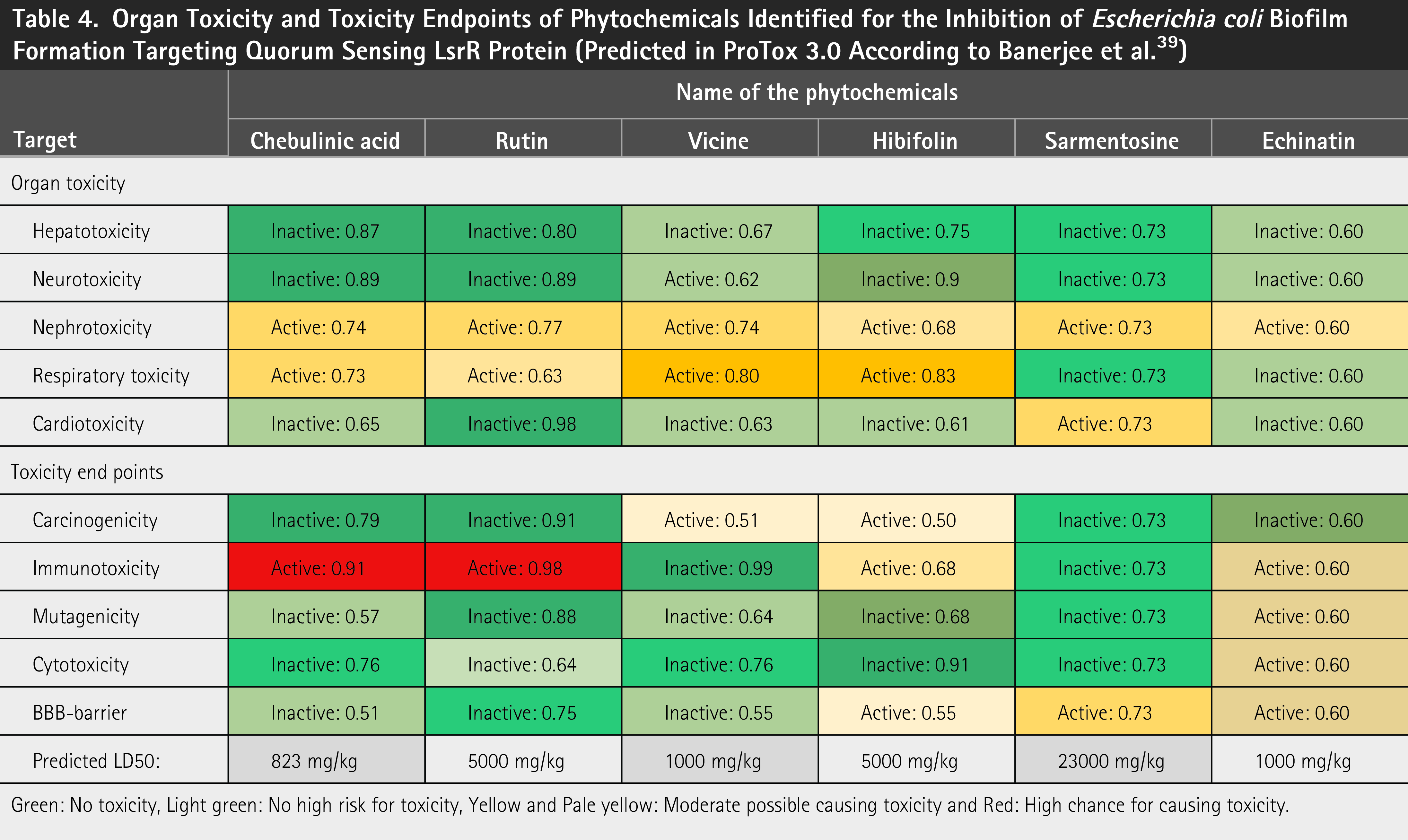
Green: No toxicity, Light green: No high risk for toxicity, Yellow and Pale yellow: Moderate possible causing toxicity and Red: High chance for causing toxicity.
CONCLUSIONS
In this study, SBVS and MD simulations identified several plant-derived bioactive compounds, including chebulinic acid, rutin, vicine, hibifolin, and sarmentosine, as potential inhibitors of the QS regulator LsrR in E. coli. Molecular docking and 100 ns MD simulations indicated stable interactions with key residues in the LsrR ligand-binding pocket, suggesting the ability of these compounds to maintain LsrR in its DNA-bound repressor conformation and potentially hinder biofilm formation. Toxicity predictions indicated that vicine, hibifolin, and sarmentosine exhibit a favorable safety profile, with low probabilities of hepatotoxicity, neurotoxicity, nephrotoxicity, cardiotoxicity, and other adverse endpoints. Chebulinic acid and rutin displayed moderate probabilities of immunotoxicity, warranting further investigation. Consequently, these findings are preliminary scaffold-level insights. Future studies incorporating physicochemical optimization, in vitro antibacterial assays, biofilm inhibition tests, and detailed toxicity profiling are essential to validate the therapeutic potential of these compounds as LsrR inhibitors and anti-QS agents to combat biofilm-mediated AMR in pathogenic bacteria.
AUTHORS’ CONTRIBUTIONS
S.V.P.: Conceptualization, investigation, methodology, data validation, review and editing, and supervision. S.S.: Investigation, formal analysis, data curation, and resources. A.C.: conceptualization, methodology, and resources. P.B.: Conceptualization, data curation, formal analysis, and validation. S.P.P.: Investigation, methodology, and formal analysis. T.P.: Conceptualization, methodology, formal analysis, and writing—review and editing. M.A.: Data curation, writing review, and editing. R.M.: Data collection, data curation, resources, and software. S.P.: Investigation, methodology, conceptualization, data curation, data validation, supervision, project administration, writing original draft, and writing—review and editing. All authors read and approved the final article.
Footnotes
DATA AVAILABILITY STATEMENT
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
AUTHOR DISCLOSURE STATEMENT
The authors declare no conflicts of interest.
FUNDING INFORMATION
This work was financially supported by Rajagiri College of Social Sciences (Autonomous), Kerala, India, under Seed Money/Faculty Minor Research (grant no. RCSS/IQAC/BB-S55/2025/155).
Supplemental Material
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.