
Abstract
Patients often suffer from hypertension, hyperlipidemia, and diabetes, requiring long-term polypharmacy. These drugs may alter gut microbiota composition, leading to adverse effects, but the underlying mechanisms are unclear. We selected 23 common drugs from ChEMBL, identified their metabolites via DrugBank, and used TargetNet and GeneCardsSuite to find drug targets and human proteins. Cross-analysis revealed 15 shared human side-effect protein targets. Molecular docking and Structure–activity relationship analyses assessed interactions with human and bacterial proteins. Eight compounds, including metabolites of amlodipine, metoprolol, glipizide, and fenofibrate, showed significant binding to human and gut bacterial proteins. Based on our in silico models, we predict that specific structural modifications could potentially enhance efficacy while minimizing microbial disruption and adverse reactions. Molecular dynamics simulations suggest that these modified drugs might computationally maintain therapeutic interactions while reducing gut microbiota disruption. Our findings highlight the impact of these drugs on gut microbiota and side effects via shared protein targets. Supported by in silico models and preliminary in vitro assays, we think that precision medicine, probiotic co-administration, or drug structure modifications could potentially serve as future strategies to mitigate complications and side effects in elderly patients, warranting further in vivo and clinical validation.
INTRODUCTION
Patients have a high prevalence of hypertension, hyperlipidemia, and diabetes due to age-related metabolic decline.1,2 These interrelated conditions significantly increase the risk of cardiovascular and neurodegenerative diseases, making long-term pharmacological treatment with antihypertensive, lipid-lowering, and antidiabetic drugs essential.3,4 While polypharmacy offers therapeutic benefits, emerging evidence suggests that chronic use of these medications may disrupt gut microbiota composition and function.5,6 Many drugs are metabolized and excreted through fecal and renal pathways, positioning the gut as a key site of drug–microbiome interaction. These interactions can lead to microbiota imbalances, reducing drug efficacy and contributing to metabolic, immune, and neurological dysfunctions, including links to diseases such as Alzheimer’s and Parkinson’s.7,8
This study adopts an integrative bioinformatics and molecular modeling approach to investigate the impact of 23 commonly prescribed “HHD Drugs” drugs on gut microbiota. Drug metabolites were structurally profiled, and reverse target screening (TargetNet and GeneCardsSuite) identified 15 human side effect proteins of interest. Molecular docking revealed significant interactions between metabolites of four drugs—amlodipine, metoprolol, glipizide, and fenofibrate—and six key bacterial proteins and seven side effect human proteins. Structure–activity relationship analysis further elucidated interaction mechanisms, informing structural optimization strategies to preserve therapeutic efficacy while minimizing microbiome disruption and clarifying the underlying molecular basis of these drugs’ side effects in humans. Crucially, our in vitro inhibition assays directly validated these computational predictions, explicitly demonstrating that derivatives of metoprolol, amlodipine, and fenofibrate directly interact with and significantly inhibit Endo-acting Polysaccharide Lyase (EPL), a key gut bacterial enzyme. This direct biological evidence strongly supports the mechanisms predicted by our in silico models. These findings provide new insights into microbiome-related effects of polypharmacy in the elderly and highlight the need for microbiome-informed precision medicine.
MATERIALS AND METHODS
Virtual Screening
Commonly used antihypertensive, lipid-lowering, and antidiabetic drugs
We extracted metabolic pathways and metabolites from DrugBank, PubChem, and literature, converting them into SMILES format for computational analysis. In addition, we explored how drug metabolites might influence gut microbiota and cause human side effects, providing a basis for further computational modeling, including reverse target identification and molecular docking.
Drug’s potential impact on gut microbiota through metabolites
To identify metabolic byproducts of commonly prescribed antihypertensives, lipid-lowering, and antidiabetic drugs, we queried the ChEMBL database by therapeutic classification and cross-referenced the results with DrugBank for metabolic pathway details. We validated the chemical structures and transformations of these metabolites using PubChem and KEGG, resolving discrepancies through pharmacokinetic studies.
Identification of common target proteins between drug metabolites and gut microbiota using target prediction and database integration
We used the TargetNet tool to predict potential target proteins for drug metabolites by submitting their SMILES representations. For gut microbiota-associated proteins, we accessed the GeneCardsSuite database and standardized the dataset by converting protein identifiers to UniProt IDs for accurate cross-referencing with predicted metabolite targets.
The two datasets were compared through UniProt ID matching to identify overlapping proteins, revealing shared targets.
Analysis of gut microbiota-associated target proteins using the RCSB PDB database
We searched the RCSB PDB using UniProt IDs, SMILES of the metabolites derived from antihypertensive, antihyperlipidemic, and antidiabetic drugs, and microbiota-related keywords to identify human side effects and structural features of gut microbiota-associated proteins targeted by drug metabolites.
Proteins with direct microbial roles were prioritized, while those involved in host metabolism and microbial signaling were also included due to their relevance in drug–microbiota interactions.
All selected proteins were annotated and analyzed for structural features and potential binding interactions with drug metabolites.9–12
Identification of potential gut microbiota targets through molecular docking and reverse target screening
We used Discovery Studio 2019 for molecular docking to explore interactions between drug metabolites and gut microbiota proteins and human side effect proteins. The process included preparing ligands by converting SMILES representations into 3D structures, energy-minimizing with CHARMM, and adjusting protonation states. High-resolution PDB structures of gut microbiota-associated proteins and human side effect proteins were retrieved, and binding sites were predicted and validated.
Molecular docking was performed using CDOCKER, with random initial poses and 10,000 sampling iterations per metabolite–protein pair. Binding energies lower than −6.0 kcal/mol were considered biologically relevant. The Ligand Interaction Analysis tool was used to analyze interactions, including hydrogen bonding and hydrophobic contacts.
To reduce false positives, we filtered proteins that docked with at least 10 distinct metabolites, prioritizing consistent interactions.13–18
Molecular dynamics simulations
Molecular dynamics (MD) simulations were performed to evaluate the stability and interaction dynamics of the designed compounds with their respective protein targets. The simulations were conducted using the GROMACS software package (version 2021.4) with the AMBER force field (ff14SB) for protein and ligand interactions. The protein-ligand complexes were solvated in a cubic box with TIP3P water molecules, ensuring a minimum distance of 1.0 nm between the solute and the box edge. Sodium and chloride ions were added to neutralize the system, and additional ions were included to achieve a physiological salt concentration of 0.15 M.
The systems were energy-minimized using the steepest descent algorithm, followed by equilibration in two stages: 1 a 100-ps NVT ensemble equilibration at 300 K using the velocity rescaling thermostat, and 2 a 100-ps NPT ensemble equilibration at 1 bar using the Parrinello-Rahman barostat. Production runs were carried out for 100 ns with a time step of 2 fs. The temperature was maintained at 300 K using the velocity rescaling thermostat, and the pressure was controlled at 1 bar using the Parrinello-Rahman barostat. The Particle Mesh Ewald method was employed to handle long-range electrostatic interactions.
The root-mean-square deviation (RMSD) was calculated to assess the stability of the ligand within the protein binding pocket over the simulation time. The RMSD values were computed for the ligand atoms relative to their initial positions after alignment of the protein backbone atoms. The RMSD analysis was performed using the GROMACS tool gmx rms with a 100-ps running average to smooth the data. The RMSD values were plotted over the simulation time to visualize the fluctuations and overall stability of the ligand-protein complexes.
In vitro inhibition assay
EPL was obtained as a recombinant protein (purity > 95%) and stored at −20°C. The glycosaminoglycan (GAG) substrate (sodium salt) was purchased from Sigma-Aldrich and prepared as a 10 mM stock solution in deionized water. Metoprolol derivative, amlodipine derivative, and fenofibrate derivative were synthesized in-house, and their purities were confirmed to be >98% by HPLC. The assay buffer consisted of 50 mM Tris-HCl, pH 7.4, containing 150 mM NaCl, 1 mM CaCl2, and 0.1% BSA. DMSO was used as the solvent for the drug derivatives.
The inhibitory activity of the drug derivatives against EPL was evaluated using a spectrophotometric assay. The reaction mixture (200 μL) contained 100 μL of assay buffer, 50 μL of GAG substrate (1 mM), 25 μL of EPL solution (0.1 mg/mL), and 25 μL of drug derivative solution at various concentrations (0.1–200 μM) or DMSO (for control). The reaction was initiated by adding the enzyme solution and incubated at 37°C for 30 min. The reaction was terminated by adding 50 μL of 1M EDTA. The release of degradation products was monitored at 235 nm using a microplate reader. The percentage of inhibition was calculated by comparing the absorbance of test samples with that of the control. The IC50 values were determined by fitting the dose-response curves to a non-linear regression model and are presented as the mean ± SD from three independent experiments performed in triplicate.
RESULTS
Virtual Screening
Commonly used antihypertensive, lipid-lowering, and antidiabetic drugs
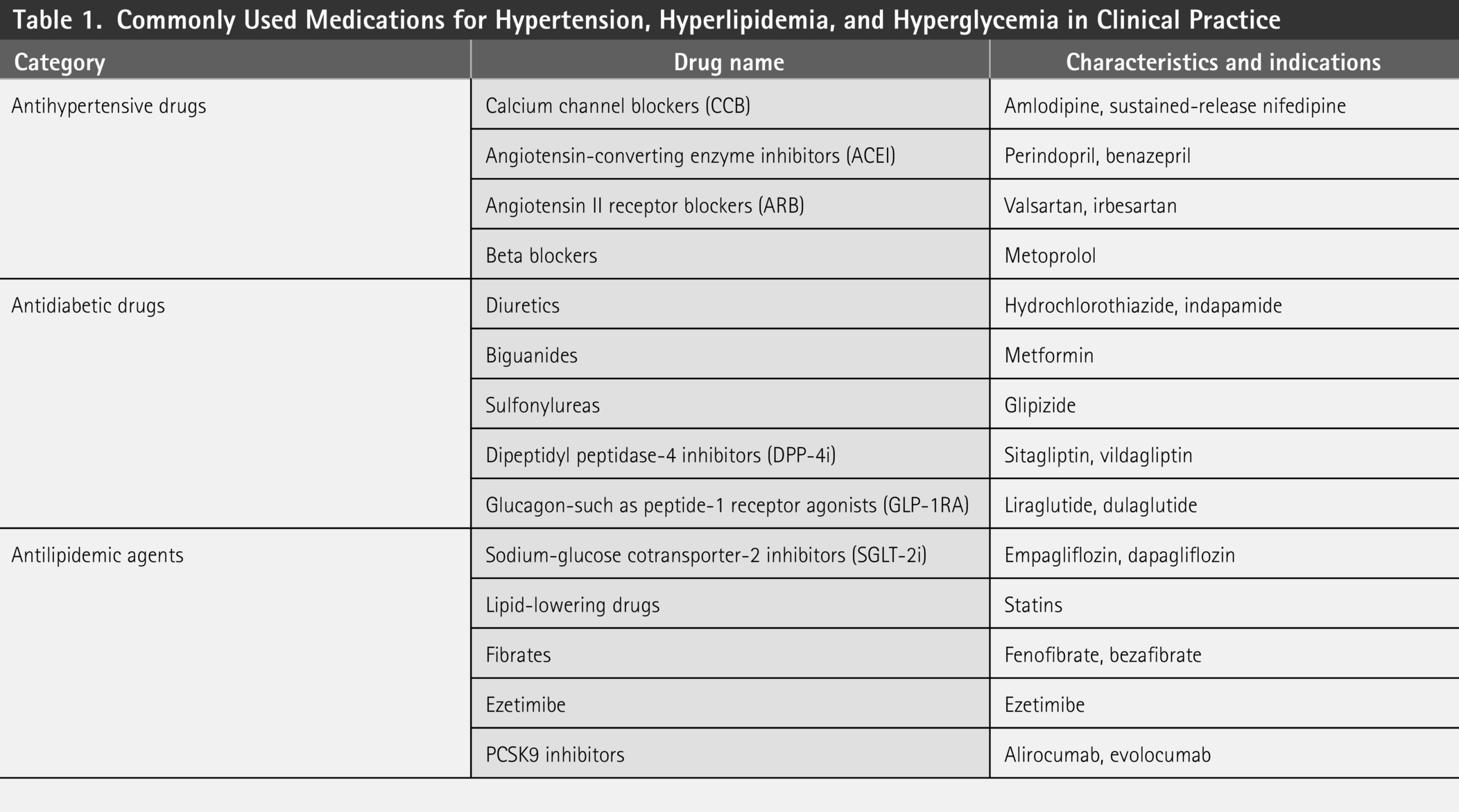
To identify commonly prescribed medications for hypertension, we focused on three therapeutic categories: antihypertensives (ACE inhibitors, ARBs, calcium channel blockers, β-blockers), lipid-lowering drugs (statins), and antidiabetics (metformin, sulfonylureas, SGLT2 inhibitors).
By reviewing the ChEMBL database, we identified several commonly prescribed antihypertensive, lipid-lowering, and antidiabetic drugs (Table 1). These drugs are widely used to manage metabolic disorders, but their metabolic byproducts may interact with the gut microbiota and human side effect proteins.
Commonly Used Medications for Hypertension, Hyperlipidemia, and Hyperglycemia in Clinical Practice
Drug’s potential impact on gut microbiota through metabolites
By consulting the DrugBank database and reviewing the names of the selected drugs, we carefully examined their metabolic pathways and identified their metabolites in the gut. We converted these metabolites into SMILES format to facilitate a more comprehensive presentation, as shown in Supplementary Table S1. This approach allows for a clearer representation of the chemical structures of the metabolites and provides a foundation for further analysis of their interactions with the gut microbiota.
Identification of common target proteins between drug metabolites and human side effect proteins using target prediction and database integration
We submitted these metabolites to the TargetNet website for target prediction, resulting in a total of 623 target proteins. Simultaneously, we utilized GeneCardsSuite to download a list of 748 target proteins associated with the human side effect. By cross-referencing these two datasets using Uniprot IDs, we identified the common targets, resulting in a set of 15 shared target proteins, as summarized in Supplementary Table S2. These specific human targets were chosen because of their critical roles in drug metabolism, systemic signaling, and transport. Functionally, they can be categorized into highly conserved metabolic enzymes (e.g., CYP1A2, CYP3A4), membrane transporters (e.g., MDR1/ABCB1, ABCG2), and key signaling proteins and receptors (e.g., PPARγ, CaSR, FFAR1). Drug interactions with these human targets can alter host metabolic and immune microenvironments, thereby indirectly perturbing the gut microbiota. This intersection provides valuable insights into the potential molecular interactions between drug metabolites and human side effect targets.
Analysis of human side effect-associated target proteins using the RCSB PDB database
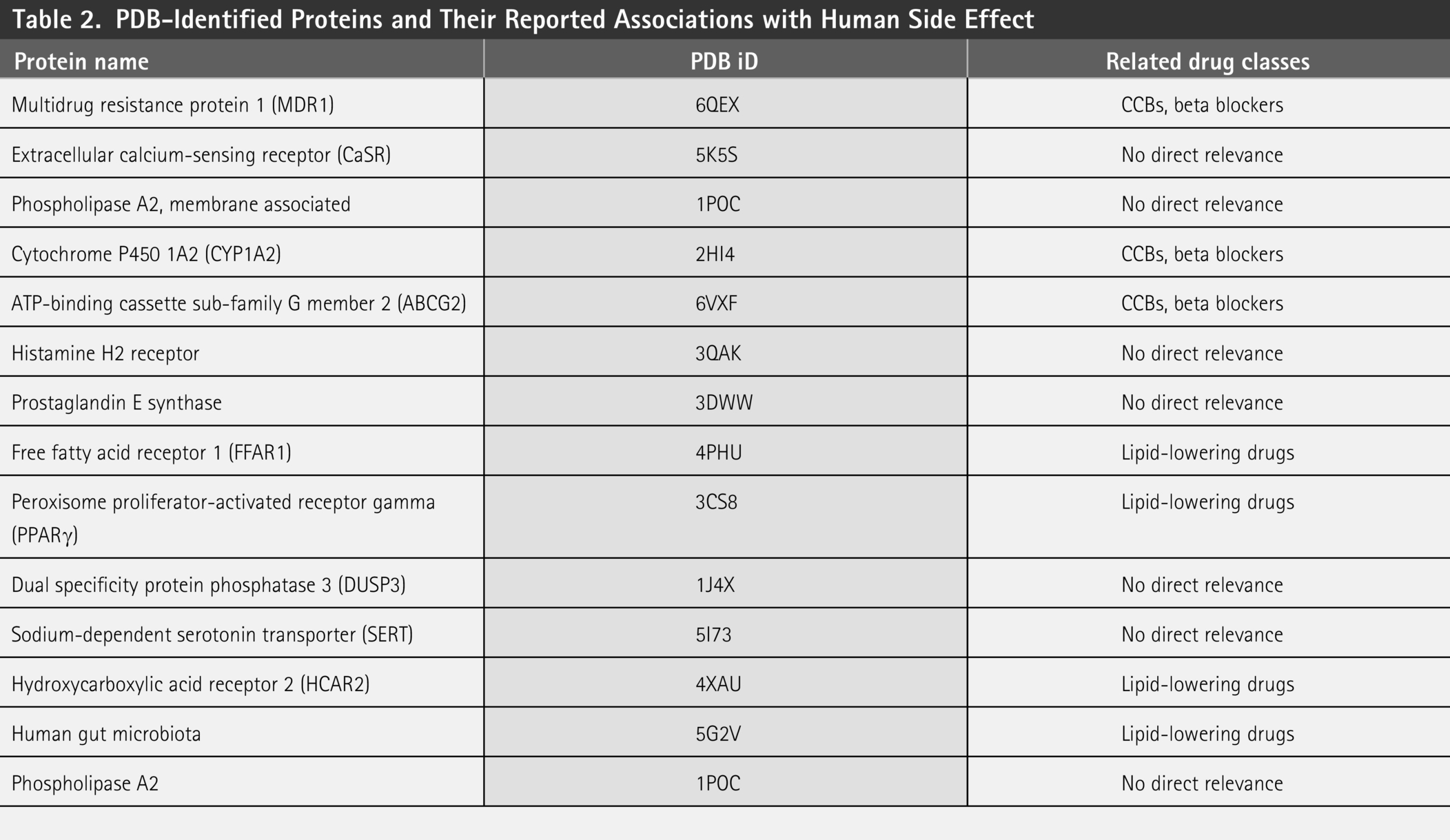
We searched the RCSB PDB database to explore protein targets linked to human side effect proteins and their potential interactions with drug metabolites in Table 2. Cytochrome P450 enzymes (CYP1A2 and CYP3A4) are essential in drug metabolism and can modulate human side effect proteins by affecting drug metabolite bioavailability.
PDB-Identified Proteins and Their Reported Associations with Human Side Effect
Lipid-lowering drugs, such as statins, primarily interact with receptors such as PPARγ, FFAR1, and HCAR2, which regulate lipid metabolism and inflammation. These receptors influence host metabolism and indirectly affect gut microbiota through metabolic signaling changes. The interactions between drug metabolites and gut microbiota proteins may alter microbial composition, impacting the efficacy and safety of long-term pharmacotherapy, particularly in elderly patients.
Identification of potential human side effect targets through molecular docking and reverse target screening
We used custom Python scripts to analyze molecular docking log files and identified seven key target proteins with consistent interaction patterns across multiple compounds (Supplementary Table S3). These proteins include the Extracellular Calcium-Sensing Receptor (CaSR) (PDB: 5K5S), which regulates calcium-dependent microbial processes; membrane-associated phospholipase A2 (1POC), influencing lipid metabolism and microbial interactions; The selected proteins—CYP1A2, H2R, FFAR1, PPARγ, and DUSP3—are closely associated with drug metabolism, immune modulation, and metabolic regulation, and their interaction with drug metabolites may underlie various adverse effects. CYP1A2, a key hepatic enzyme, influences drug clearance and toxicity, with its inhibition potentially leading to hepatotoxicity and drug–drug interactions. H2R plays a central role in gastric acid secretion, and its dysregulation may contribute to gastrointestinal disturbances and electrolyte imbalance. FFAR1 modulates insulin secretion in response to fatty acids, and prolonged activation may increase the risk of hypoglycemia and β-cell dysfunction. PPARγ, a nuclear receptor targeted by antidiabetic drugs, is linked to fluid retention, weight gain, cardiovascular risks, and skeletal fragility. DUSP3 is implicated in MAPK pathway regulation and inflammatory responses; its modulation by microbial metabolites may contribute to immune imbalance and metabolic disorders. Collectively, these findings highlight the potential mechanisms by which gut microbiota–drug interactions contribute to human side effects, particularly in elderly patients undergoing long-term therapy.
Furthermore, to validate our findings, we performed reverse target screening by cross-referencing the docked metabolites identified earlier. This involved UniProt ID mapping and intersection analysis using Venny 2.1, resulting in a refined dataset labeled “Abundon1” (Figs. 1 to 2), which captures the most biologically relevant drug–metabolite–microbiota interactions. This dataset provides deeper insights into how specific drug metabolites interact with human side effect proteins, which could inform therapeutic strategies.
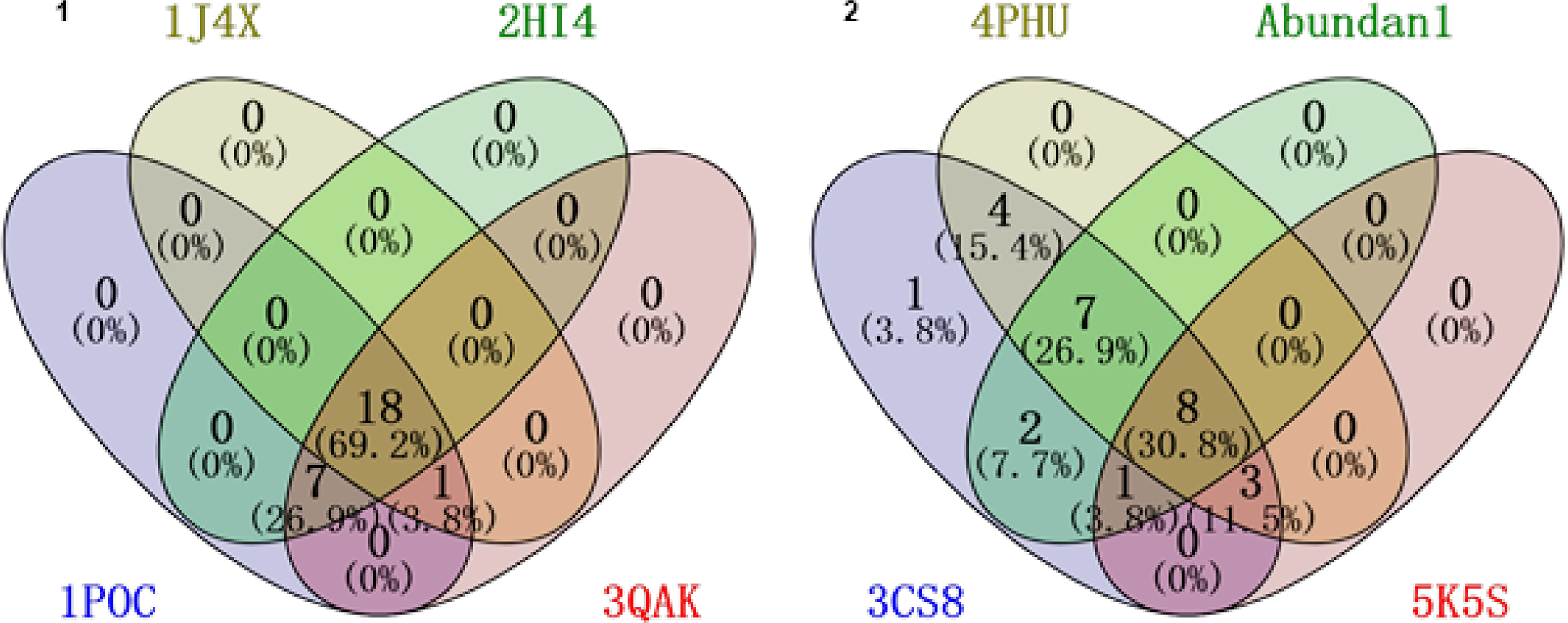
Predicted target proteins from drug metabolite docking analysis.
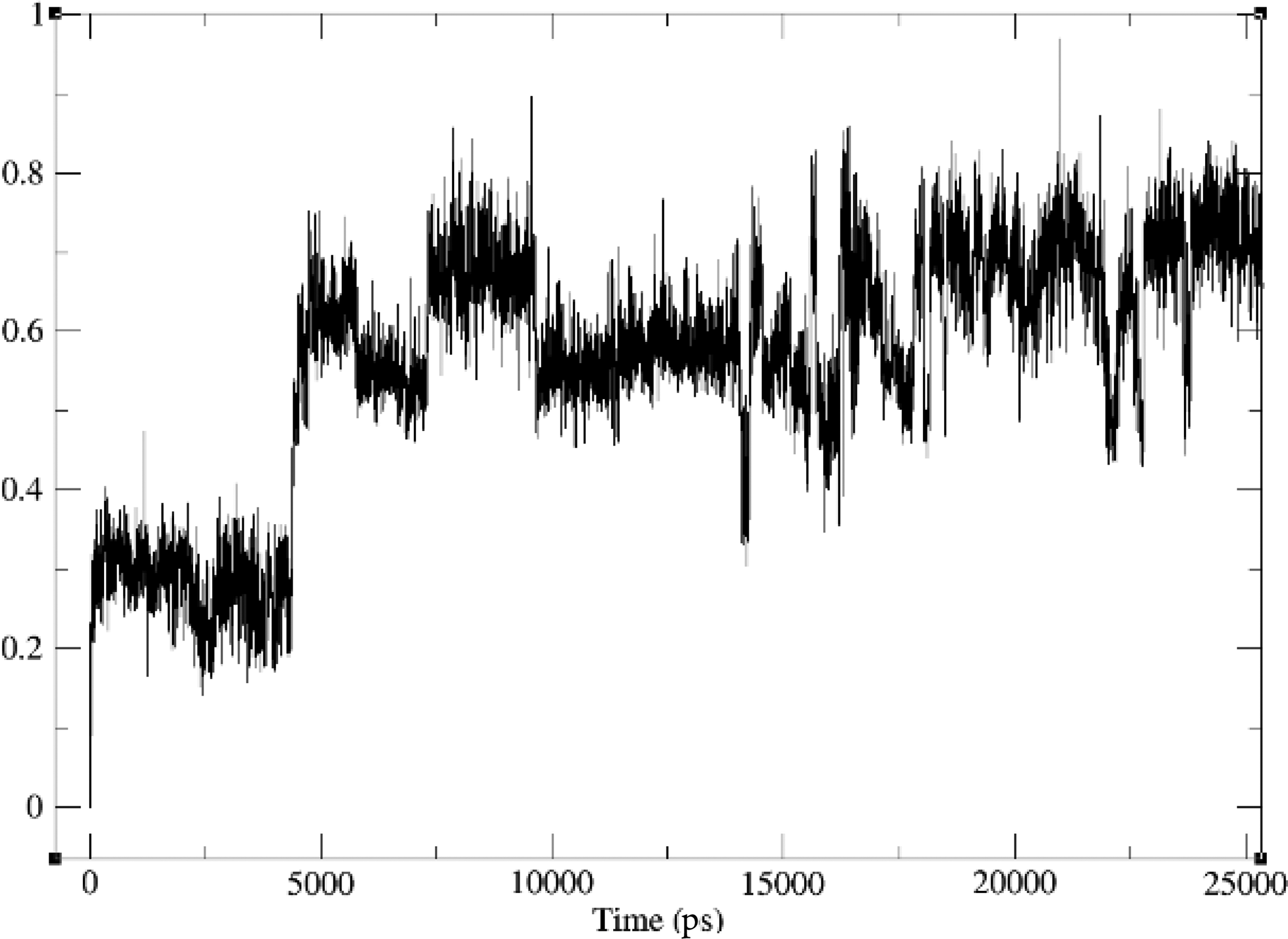
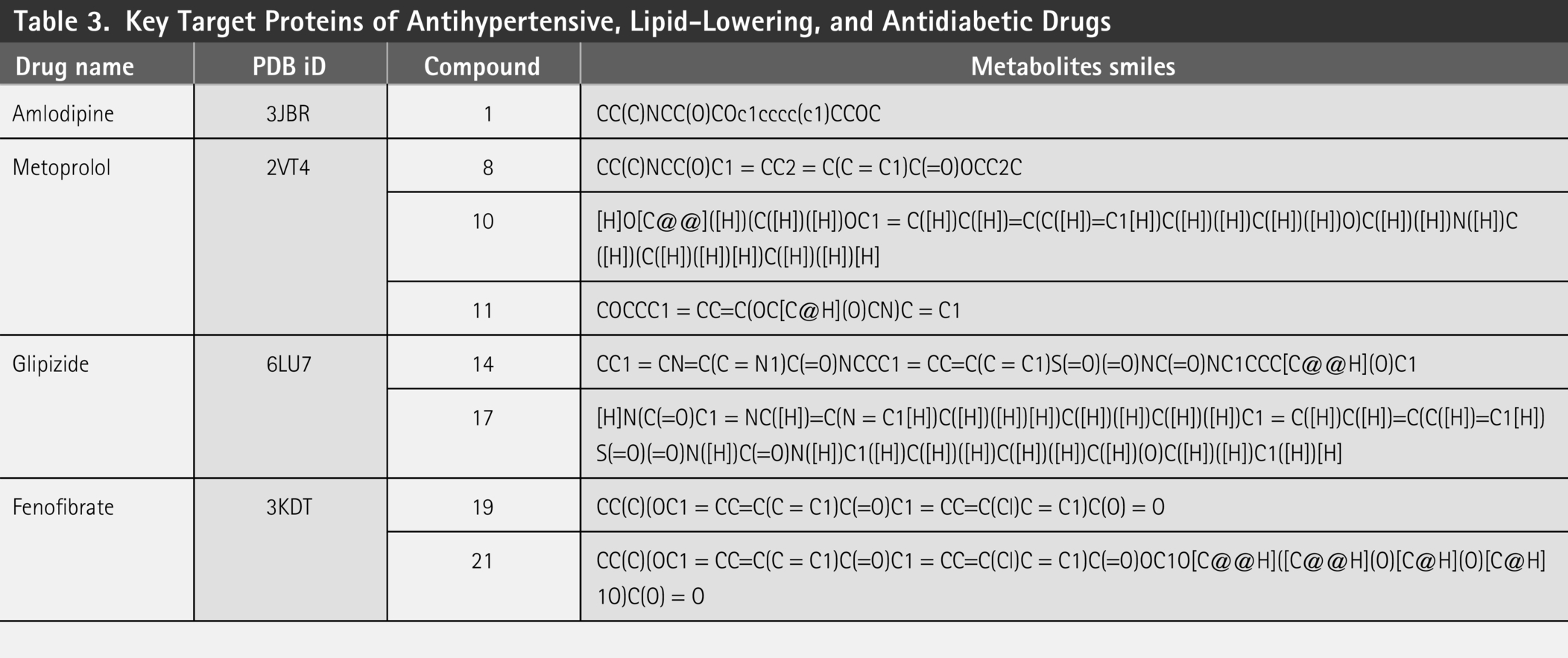
RMSD of metoprolol derivative with target protein (5G2V). RMSD, root-mean-square deviation.
Our analysis also identified metabolites from amlodipine, metoprolol, glipizide, and fenofibrate (Table 3). Notably, glipizide metabolites (Compounds 14 and 17) are primarily excreted via the kidneys and do not affect gut microbiota composition. These findings support our predicted drug–metabolite–microbiota interactions and emphasize the importance of considering microbiome-related effects in drug therapy design, potentially enhancing drug efficacy and reducing adverse reactions.
Key Target Proteins of Antihypertensive, Lipid-Lowering, and Antidiabetic Drugs
MD simulations and impact on gut microbiota
We employed MD simulations to investigate the interactions between three drug derivatives and their respective protein targets. The analysis revealed that these compounds demonstrated satisfactory RMSD values, indicating stable binding interactions with the target proteins. Specifically, the metoprolol derivative exhibited an RMSD of approximately 0.6 nm when bound to the human target protein (PDB ID: 5G2V), as shown in Figure 2. Similarly, the amlodipine derivative displayed an RMSD of around 0.75 nm with the same human target protein (PDB ID: 5G2V), depicted in Figure 3. In addition, the Fenofibrate derivative achieved an RMSD of about 0.25 nm when interacting with the gut bacterial target protein (PDB ID: 5G2V), illustrated in Figure 4. These results suggest that our modification strategy effectively maintained the therapeutic efficacy of the drugs while significantly minimizing their adverse effects on the gut microbiota.
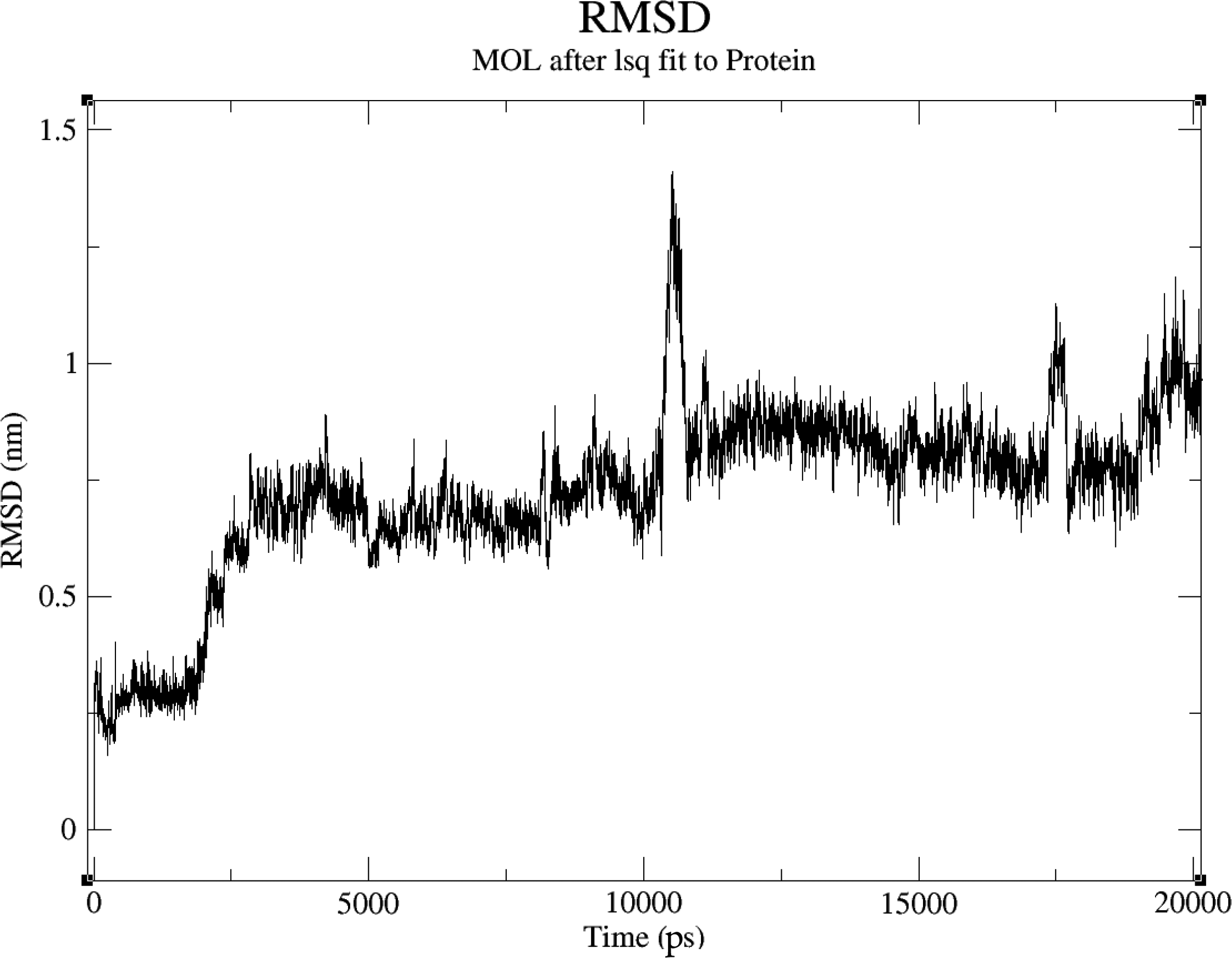
RMSD of amlodipine derivative with target protein (5G2V). RMSD, root-mean-square deviation.
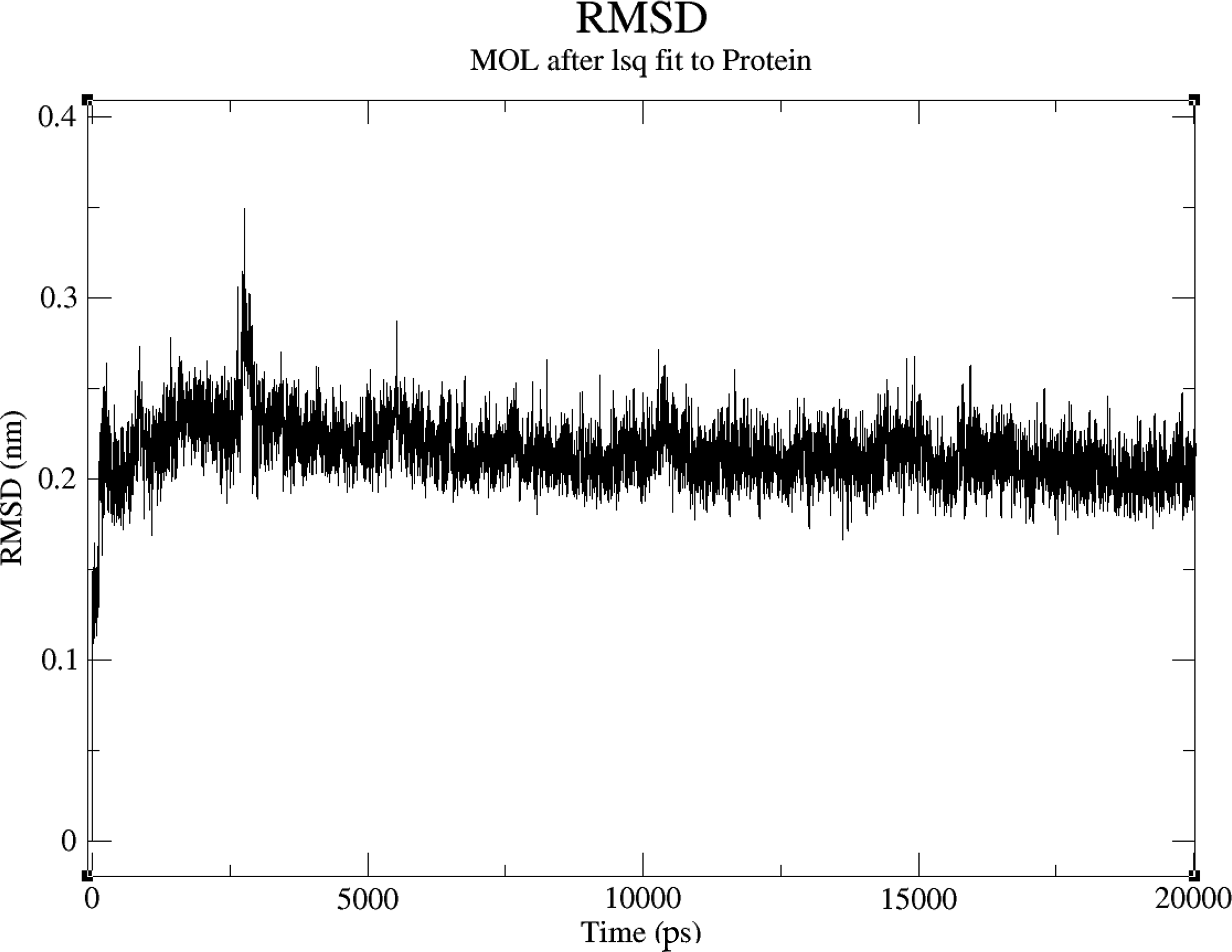
RMSD of fenofibrate derivative with gut bacterial target protein (5G2V). RMSD, root-mean-square deviation.
In vitro activity
As presented in Table 4, we conducted in vitro activity assays to evaluate the inhibitory effects of the three drug derivatives on EPL, an enzyme involved in the degradation of GAGs. The results demonstrated that all three compounds exhibited substantial inhibitory activities against EPL, with IC50 values of 105.0 μM for the metoprolol derivative, 199.4 μM for the amlodipine derivative, and 12.0 μM for the fenofibrate derivative. Given that GAGs represent a critical nutritional source for certain members of the gut microbiota, inhibition of EPL is likely to directly impair the ability of bacteria to degrade these polysaccharides. Consequently, this inhibition may exert a significant impact on bacterial strains that rely on GAGs as a primary nutrient source.
Inhibition of Endo-Acting Polysaccharide Lyase by Three Drugs
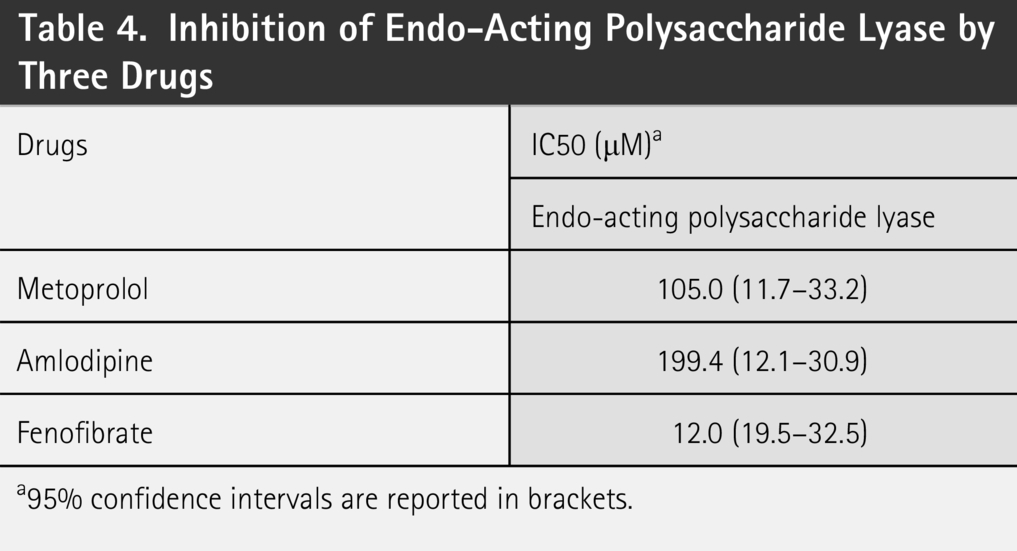
a95% confidence intervals are reported in brackets.
Comparative analysis of drug-target and off-target interactions of amlodipine, metoprolol, and fenofibrate: strategies for structure-based optimization
After reviewing the literature, we found that amlodipine, 19 metoprolol, 19 and fenofibrate 20 exhibit strong inhibitory effects on the gut microbiota. To mitigate the impact of amlodipine, metoprolol, and fenofibrate on the gut microbiota while maintaining their therapeutic efficacy, we implemented targeted structural modifications based on their molecular interaction profiles in Figure 5. For Metoprolol, the phenethyl ether group, which contributed to off-target binding with human proteins, was either removed or modified by introducing less polar functional groups to disrupt π–π stacking interactions. In addition, the overall polarity of the molecule was reduced to minimize unintended bacterial interactions. For amlodipine, the hydroxyl and aromatic groups were modified by replacing the hydroxyl with a fluorine atom and introducing electron-withdrawing groups to the aromatic ring to reduce off-target binding. The lactone ring and benzyl alcohol/ether groups in amlodipine metabolites were also altered to reduce electronegativity and eliminate benzyl structures, thereby minimizing off-target bacterial interactions. For fenofibrate, the carboxyl group was esterified, and less polar functional groups were introduced to reduce binding affinity with off-target proteins. Bulky substituents and electron-withdrawing groups were added to the aromatic rings to hinder π–π stacking interactions with bacterial proteins. These modifications were designed to enhance the selectivity of the drugs for their primary targets while reducing off-target interactions with both human and bacterial proteins. The detailed information on these modifications is provided in SI 1.4.
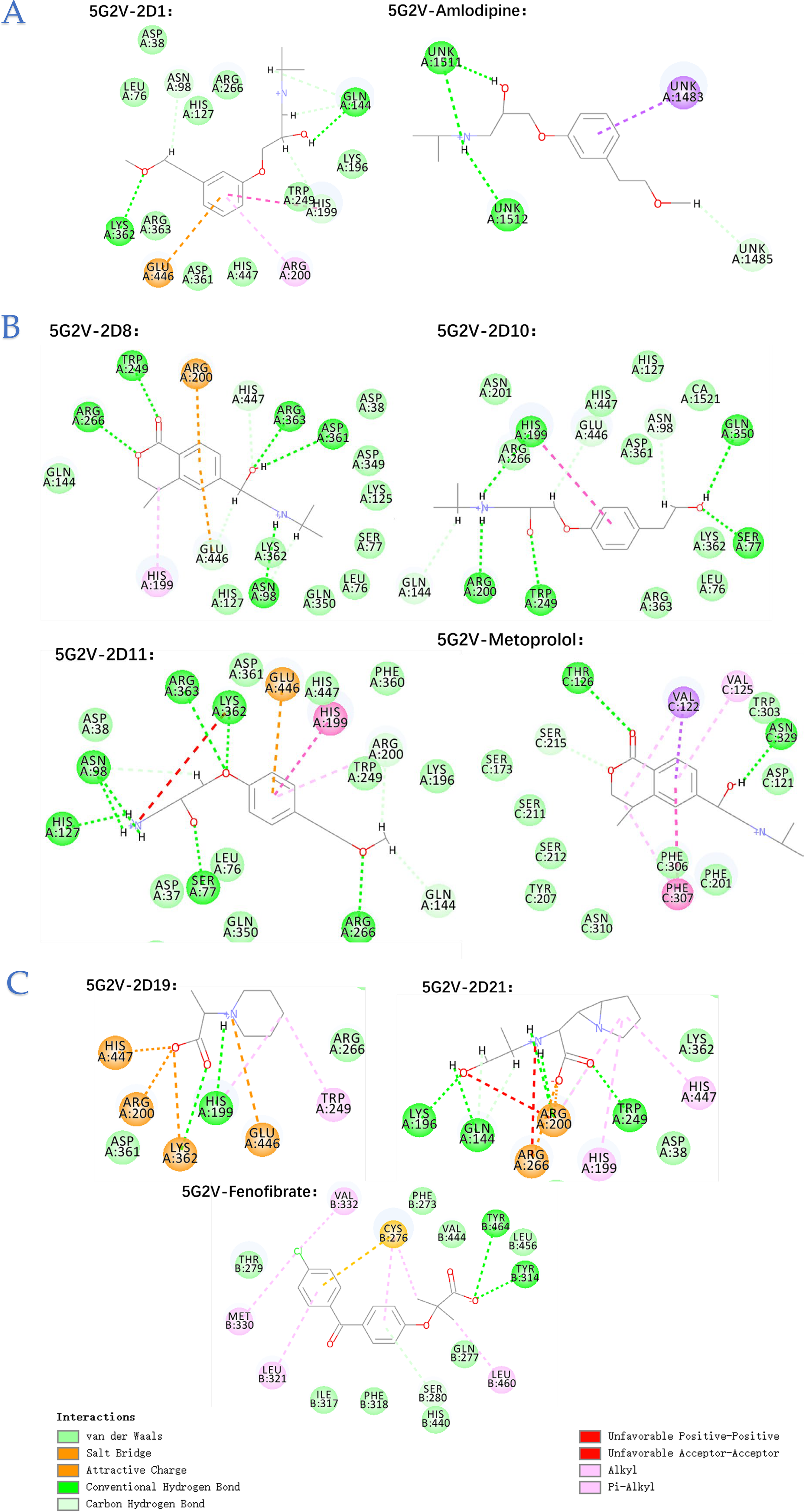
Molecular docking interactions of gut metabolites with endo-acting polysaccharide lyase (5G2V) and drug targets.
CONCLUSIONS
Our study has provided a comprehensive analysis of the interactions between commonly prescribed antihypertensive, lipid-lowering, and antidiabetic drugs and the gut microbiota. Through virtual screening, molecular docking, and dynamics simulations, we identified key metabolites and their potential impact on human side effect proteins and gut microbiota. The findings highlight that specific proteins such as CYP1A2, H2R, FFAR1, PPARy, and DUSP3 play crucial roles in mediating adverse effects, and our models predict that targeted structural modifications of drugs such as amlodipine, metoprolol, and fenofibrate can enhance their selectivity while reducing off-target interactions. most importantly, our in vitro activity assays provided direct biological evidence of these interactions, confirming that these drug derivatives directly and substantially inhibit the bacterial enzyme EPL. These proposed modifications, including altering functional groups and introducing electron-withdrawing groups, are computationally predicted to maintain therapeutic efficacy while minimizing adverse effects on the gut microbiota.
In summary, our combined computational and in vitro research underscores the importance of considering gut microbiota–drug interactions in the design of antihypertensive, lipid-lowering, and antidiabetic therapies. Based on these findings, we think that optimizing these interactions could potentially enhance drug efficacy and safety, particularly for elderly patients undergoing long-term pharmacotherapy. Furthermore, beyond structural modifications, recent advancements in drug delivery systems suggest that reformulating antihypertensive and antidiabetic drugs with microbiome-promoting agents 21 (e.g., targeted release to avoid upper GI disruption or co-formulation with prebiotics) represents an emerging and highly viable strategy to preserve gut homeostasis. However, it is important to note that while our in silico and in vitro results provide a strong mechanistic foundation, proposed clinical applications such as microbiome-informed precision medicine or probiotic co-administration remain explicitly as hypotheses at this stage. Future work must rigorously focus on validating these findings through in vivo animal models and clinical trials to translate these optimization strategies into true clinical practice.
Despite the insights gained from our combined computational and in vitro approach, this study has several limitations that must be acknowledged. Primarily, while the in vitro EPL inhibition assay provides direct biological evidence of specific drug–enzyme interactions, it does not fully capture the complex, multifaceted, and dynamic nature of the in vivo gut microbiome ecosystem. The translation from the inhibition of a single bacterial enzyme to the functional disruption of the entire microbial community is complex and requires further investigation. More in-depth experimental work, including in vivo animal models, multi-omics microbiome profiling, and rigorous clinical trials, is absolutely essential to validate these mechanisms and fully elucidate the ecological and clinical impact of these polypharmacy interactions.
INSTITUTIONAL REVIEW BOARD STATEMENT
Although this paper involves the development of antibiotics, animals and humans are not involved in the experimental process, so no ethical review and approval are required.
INFORMED CONSENT STATEMENT
Informed consent was obtained from all subjects involved in the study.
DATA AVAILABILITY STATEMENT
Data are provided within the article or Supplementary Data files.
AUTHORS’ CONTRIBUTIONS
Conceptualization, N.X. and Y.Z.; methodology, N.X. and Y.Z.; software, N.X. and Y.Z.; validation, N.X. and Y.Z.; formal analysis, N.X. and Y.Z.; investigation, N.X. and Y.Z.; resources, N.X.; data curation, N.X. and Y.Z.; writing—original draft preparation, N.X.; writing—review and editing, N.X. and Y.Z.; visualization, N.X. and Y.Z.; supervision, N.X. and Y.Z.; project administration, N.X. and Y.Z.; funding acquisition, N.X. and Y.Z.
Footnotes
DISCLOSURE STATEMENT
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
FUNDING INFORMATION
This review was supported by Cangzhou City Science and Technology Program Self-Funded Project (NO : 222106046).
Supplemental Material
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.