
Abstract
Odontogenic keratocysts are relatively common lesions that may occur in isolation or in association with nevoid basal cell carcinoma syndrome (or Gorlin syndrome). The PTCH gene has been reported to be associated with Gorlin syndrome. We investigated 10 cases of non-syndromic keratocysts and two other cases associated with Gorlin syndrome, looking for PTCH mutations. Four novel and 1 known PTCH mutations were identified in five individual patients. Of the 5 mutations identified, 2 were germ-line mutations (2619C>A; 1338_1339insGCG) in 2 cysts associated with Gorlin syndrome, and 3 were somatic mutations (3124_3129dupGTGTGC; 1361_1364delGTCT; 3913G>T) in 3 non-syndromic cysts. This report describes PTCH mutations in both non-syndromic and Gorlin-syndrome-related odontogenic keratocysts in Chinese patients, and suggests that defects of PTCH are associated with the pathogenesis of syndromic as well as a subset of non-syndromic keratocysts.
INTRODUCTION
The odontogenic keratocyst is an aggressive cystic lesion that has a putative growth potential and a propensity for recurrence (Browne, 1971; Li et al., 1994). Although the great majority of keratocysts occur in isolation as single, non-syndromic cysts, they may also present as multiple cysts as a feature of the nevoid basal cell carcinoma syndrome (Gorlin snydrome, OMIM#109400). Gorlin syndrome is a rare autosomal-dominant disorder with variable clinical manifestations, such as basal cell carcinoma of the skin, keratocysts of the jaws, palmar or plantar pits, ectopic calcification of the falx cerebri, etc. (Gorlin, 1987; Kimonis et al., 1997). Multiple jaw keratocysts are the most consistent and common manifestation of the syndrome, occurring in 65–100% of patients (Gorlin, 1987). The syndrome-associated keratocysts are found in both jaws with equal frequencies, in contrast to non-syndromic cysts, which are most frequently associated with the lower jaw (Lo Muzio et al., 1999a). Keratocysts often represent the first manifestations of Gorlin syndrome, frequently preceding syndromic basal cell carcinomas, thus facilitating early diagnosis (Lo Muzio et al., 1999b).
It is now believed that Gorlin syndrome is caused by germ-line mutations of the PTCH gene (Hahn et al., 1996; Johnson et al., 1996). In about 60–85% of individuals fulfilling the diagnostic criteria of the syndrome, it is possible to identify the underlying PTCH defect (Evans and Farndon, GeneReview at www.genetests.org, 2004). The PTCH gene is the human homologue of the Drosophila segment polarity gene patched and has been localized to chromosome 9q22.3-q31 (GenBank accession numbers: U43148 and U59464; Hahn et al., 1996; Johnson et al., 1996). It encodes a 12-transmembranous-domain protein that physically binds at least 1 of the 3 known vertebrate Hedgehog molecules—Sonic hedgehog (SHH)—with high affinity, controlling cell fate and embryonic patterning in numerous tissues (Stone et al., 1996; Hardcastle et al., 1998). Apart from the high frequency of germ-line mutations of the PTCH gene detected in patients with the syndrome, somatic mutations of PTCH have also been identified in a range of sporadically occurring tumors, including those observed in the syndrome, i.e., basal cell carcinomas (Gailani et al., 1996; Hahn et al., 1996; Johnson et al., 1996), medulloblastoma (Xie et al., 1997), and trichoepithelioma (Vorechovsky et al., 1997). Similarly, several studies demonstrated the presence of PTCH mutations in syndromic and non-syndromic keratocysts (Lench et al., 1997; Barreto et al., 2000; Ohki et al., 2004). However, the prevalence and range of PTCH mutations in odontogenic keratocysts remain to be established, in view of the limited number of cysts examined to date. The present study aimed to analyze PTCH mutations in a group of Chinese patients presenting with non-syndromic and Gorlin-syndrome-related keratocysts.
MATERIALS & METHODS
Subjects and Samples
Samples of 12 keratocysts from 12 unrelated patients were obtained from the Department of Oral Pathology, Peking University School of Stomatology. The fresh tissue specimens were divided into two parts. The first part was fixed in 10% formalin and routinely processed for histological diagnosis. Ten lesions were classified as non-syndromic keratocysts, and 2 were from patients with Gorlin syndrome diagnosed according to established criteria (Kimonis et al., 1997; see APPENDIX). The second part of the tissue specimen was immediately frozen and stored at −80°C for subsequent PCR and sequencing analysis. Peripheral blood was also collected from all patients. Informed consent was obtained from all study participants, and the study protocol was approved by the Ethical Committee of Peking University Health Science Center.
DNA Extraction and Polymerase Chain-reaction (PCR)
Genomic DNA from frozen samples (25 mg) of cyst tissue was extracted with a DNeasy Tissue Kit (Qiagen, Valencia, CA, USA). DNA from peripheral blood was isolated with a Whole Blood Genomic DNA Mini Kit (V-gene Biotechnology Limited, Hangzhou, P.R. China). Each of the 23 exons of the PTCH gene was amplified separately with specific primers, as previously described (Chidambaram et al., 1996; Hahn et al., 1996; Xie et al., 1997), except for exon 14 and exon 23. Exon 14 was amplified in 2 pieces, 5′-AAAATGGCAGAATGAAAGCACC-3′, 5′-CTGAGGGTGTCC TGTGTCAC-3′ and 5′-CACACGCACGTGTACTACAC-3′, 5′-CTGATGAACTCCAAAGGTTCTG-3′. Exon 23 was also amplified in 2 pieces, 5′-AACCCAAGGAGGGAAGTGTG-3′, 5′-AAGCCGTCACAGTGGTGATG-3′ and 5′-TCTACTGAAGGG CATTCTGGC-3′, 5′-GAACCTTGTCCTCCTCTTTGC-3′. PCRs were performed in a final volume of 50 μL containing approximately 100 ng of template DNA, 200 μM dNTPs, 10 pmol of each primer, 1.25 u of Taq polymerase [TaKaRa Biotechnology (Dalian) Co., Ltd, P.R. China], 50 mM KCl, 10 mM Tris-HCl, and 1.5 mM MgCl2. Amplification was performed for 35 cycles at 94°C for 30 sec, 57°C for 30 sec, and 72°C for 30 sec in a thermal cycler (PTC-100; MJ Research, Watertown, MA, USA).
Direct Sequencing
PCR products were gel-purified with a Gel Extraction kit (Omega Bio-Tek, Doraville, GA, USA) , according to the manufacturer’s protocol, and directly sequenced with the same primers as for the original PCR amplification. When insertion or deletion of multiple nucleotides occurred, and direct sequencing from the PCR products became difficult, further mutation detection was pursued in a subset of samples by the cloning of purified PCR product into the plasmid vector pGEM-T (Promega, Madison, WI, USA). After transformation into the competent E. coli strain TOP10, colonies carrying recombinant plasmid were selected, and the plasmid DNA was isolated with the use of a Plasmid Miniprep Kit (Sigma, St. Louis, MO, USA). Plasmid DNA was sequenced with M13 universal forward and reverse primers. Sequencing analysis was performed on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Any mutation detected was confirmed by reverse sequencing and by analysis of samples from at least 2 independent PCRs.
RESULTS
PTCH gene mutations were examined in 10 non-syndromic and 2 syndromic keratocysts by direct sequencing. Four novel and 1 known PTCH mutations were identified in 5 cysts, 3 of which were non-syndromic cases, and 2 of which were associated with Gorlin syndrome (Table). These mutations were distributed throughout the gene. In addition, 8 previously reported polymorphisms were also found in 11 of the 12 cases (Table).
Somatic Mutations in Non-syndromic Keratocysts
Somatic mutations were identified in 3 out of 10 non-syndromic keratocysts. These 3 novel mutations were evident only in DNA isolated from the cyst tissues, and were not detected in samples from the corresponding peripheral blood. A duplication of 6 nucleotides following position 3129 in exon 18 was detected in a non-syndromic case (Fig. 1A). This mutation causes a duplicated insertion of valine and cysteine residues between codons 1043 and 1044. The second mutation involved deletion of 4 nucleotides at position 1361_1364 in exon 10 (Fig. 1B). This frameshift mutation introduces a stop codon at amino acid residue 454. Direct sequencing of exon 23 from another non-syndromic cyst revealed a G>T transversion at nucleotide 3913 (Fig. 1C). This caused a change from aspartic acid to tyrosine at codon 1305.
Germ-line Mutations in Syndromic Patients
A known nonsense mutation was detected in a 21-year-old female syndromic patient. Direct sequencing of both the cyst and peripheral blood samples revealed a C>A substitution at nucleotide 2619 in exon 16. This caused a tyrosine to a stop codon substitution at amino acid residue 873 (Fig. 2A). This patient had a family history of Gorlin syndrome and presented with multiple keratocysts, cleft lips and palate, bifid ribs, and multiple facial naevi. In addition, multiple recurrences of keratocysts in the upper and lower jaws were recorded in this patient over a period of 7 yrs. A triplet nucleotide insertion at position 1338 in exon 9 (Fig. 2B) was detected in both the jaw cyst and peripheral blood from the other syndromic patient. This mutation introduced an alanine between codon 446_447. In addition, it also created a novel restriction enzyme cleavage site for MluI (5′-ACGCGT-3′), thus allowing its presence to be confirmed by restriction-enzyme analysis (Fig. 2C). This 37-year-old male patient also had a family history of the syndrome and presented manifestations of recurrent multiple keratocysts, calcification of the falx cerebri, and multiple skin naevi.
DISCUSSION
The PTCH protein serves as a receptor for the secreted Sonic hedgehog (SHH) protein, and inhibits the signaling pathway by repressing the activity of Smoothened (SMO), another transmembranous protein (Stone et al., 1996). According to this model, PTCH inhibition of SMO is lifted upon binding of SHH to PTCH or following mutational inactivation of PTCH. The SHH signaling pathway plays an important role in mammalian embryonic development of structures such as the neural tube, axial skeleton, limbs, lungs, skin, hair follicles, and teeth (Hardcastle et al., 1998). SHH signaling also regulates growth and determines the shape of teeth (Dassule et al., 2000). Addition of exogenous SHH protein directly to tooth germs during early development may result in abnormal epithelial invagination. Recent experiments showed that at the stage of initiation of odontogenesis in the murine mandibular arch, the SHH target genes such as PTCH and GLI-1 are up-regulated in the diastema mesenchyme (Cobourne et al., 2004). Odontogenic keratocysts almost certainly arise from derivatives of the dental lamina, an embryonic structure that normally differentiates into tooth buds and enamel-producing cells during odontogenesis (Browne, 1971; Gorlin, 1987). The remnants of dental lamina usually regress at later stages of development. The precursor cells of jaw cysts may fail to involute, due to a genetic alteration. Alternatively, dental lamina remnants may remain quiescent in childhood and adult life, unless a genetic event creates a hyperproliferative clone of cells. The epithelium lining keratocysts does show higher proliferative activity than the linings of other types of jaw cysts (Li et al., 1994). The demonstration that PTCH is a tumor suppressor gene, probably responsible for Gorlin syndrome and its related sporadic neoplasms, prompted researchers to investigate the role of this gene in the pathogenesis of keratocysts. Loss of heterozygosity in the 9q22.3 region has been demonstrated with high frequency in both syndromic and non-syndromic keratocysts (Levanat et al., 1996). There have been three reports in the literature involving PTCH mutational analysis of DNA derived from keratocyst tissues (Lench et al., 1997; Barreto et al., 2000; Ohki et al., 2004). Lench et al.(1997) identified PTCH mutations in 5 out of 16 syndrome-related keratocysts. These 5 mutations were all germ-line mutations, 4 of which caused frameshifts and premature protein truncation. In a subsequent study of 3 non-syndromic and 3 syndrome-related keratocysts, PTCH mutations were found in 3 cysts, with 2 germ-line mutations associated with Gorlin syndrome and 1 somatic mutation in a non-syndromic cyst (Barreto et al., 2000). A more recent study by Ohki et al.(2004) failed to identify any PTCH mutations in 4 syndromic keratocysts, but 13 mutations were detected in 7 out of the 18 non-syndromic keratocysts. It is not clear, however, whether these identified mutations were somatic or germ-line, since the constitutional DNA of these cases was not available for analysis.
In the present study, we have identified 4 novel and 1 known PTCH mutations in 5 cysts, 2 of which were associated with Gorlin syndrome. A known germ-line mutation found in a syndrome patient was a nonsense mutation (2619C>A) resulting in PTCH protein truncation in the second extracellular loop. An identical germ-line mutation has been previously reported in a French Gorlin syndrome patient (Boutet et al., 2003). The second extracellular loop of PTCH is known to be an important domain that interacts with SHH (Gailani et al., 1996). Thus, PTCH protein truncation in this region may inactivate its ability to bind SHH ligand. The novel germ-line mutation (1338_1339insGCG) identified from the other syndrome patient resulted in an insertion of alanine in the second transmembrane domain of the PTCH protein, a region known as the sterol-sensing domain (SSD). Analysis of recent data from Drosophila suggests that the SSD may play a role in mediating intracellular PTCH trafficking as a means of regulating Smoothened (Strutt et al., 2001). Thus, insertion of an amino acid residue in the SSD may disturb signaling transduction in the SHH signaling pathway. We also identified 3 novel somatic mutations in 3 non-syndromic keratocysts. One deletion mutation (1361_1364delGTCT) resulted in a frameshift and premature protein truncation in the SSD. The other was a duplication mutation (3124_3129dupGTGTGC), which introduced 2 extra amino acid residues in the 8th transmembrane domain. The third one was a missense mutation (3913G>T), which caused exchange of an acidic amino acid (Asp) for a neutral polar amino acid (Tyr) near the C terminus of the PTCH protein. To characterize further the 4 novel mutations identified in the present study, we tested 100 unrelated control DNA samples by PCR-SSCP analysis and found that the abnormal SSCP migration bands seen in the 4 novel mutant samples were absent from the control DNAs (see APPENDIX). Therefore, these newly identified mutations are unlikely to be rare polymorphisms. Thus, analysis of our data provides further evidence that defects of PTCH are involved in the formation of syndromic as well as non-syndromic keratocysts, although further studies are now required to identify how these mutations may impair PTCH function.
Eight polymorphisms, previously reported in North American (Xie et al., 1997), European (Richards et al., 1997; Bodak et al., 1999; Boutet et al., 2003), and Japanese (Fujii et al., 2003) populations, were also identified in 11 of the 12 patients in the present series. Some of these polymorphic variants (i.e., 1686C>T, 2199A>G, 3944T>C, IVS10-51G>C, IVS10-8T>C) occurred with a high frequency, and eight patients had 2 or more polymorphisms. A recent study has reported that certain haplotypes of PTCH polymorphisms could mediate susceptibility to basal cell carcinomas (Strange et al., 2004). Further studies are needed to define such an association between susceptibility to odontogenic keratocyst formation and PTCH polymorphisms.
Summary of PTCH Mutations and Polymorphisms in Odontogenic Keratocysts
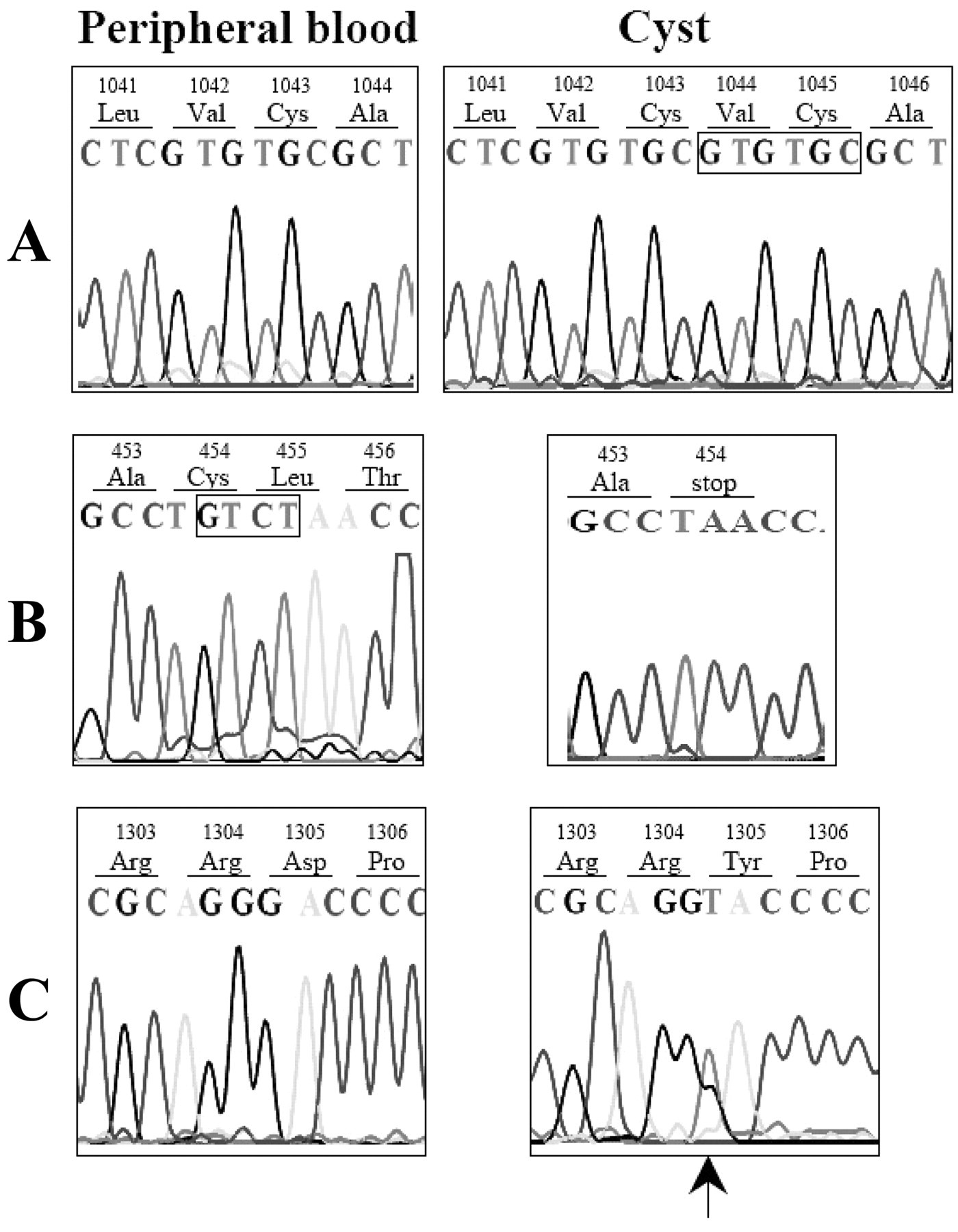
Three somatic mutations of PTCH identified in 3 non-syndromic keratocysts. Sequences of both peripheral blood (left) and cyst (right) are shown.

Two germ-line mutations of the PTCH gene identified in two patients with Gorlin syndrome
Footnotes
Acknowledgements
This work was supported by Research Grants from the National Nature Science Foundation of China (30240031 and 30572048) and the Municipal Nature Science Foundation of Beijing (7032031). The authors also thank Professor Dalong Ma, Dr. Mingxu Xu (Center for Human Disease Genomics, Peking University Health Science Center) for their meaningful discussions.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.