
Abstract
Autophagy, an evolutionarily conserved and tightly controlled process in eukaryotic cells, allows them to respond to stress by selectively eliminating dysfunctional or unwanted materials to promote metabolic flexibility and maintain homeostasis. While autophagy is orchestrated by different autophagy (ATG)-related genes, whose regulation varies considerably according to tissue type and developmental stage. In this review, we investigate how regardless of the different players involved in autophagy, ATG5 emerges as a unique, highly conserved, critical molecule that acts as a central rheostat to control the stem cell fate, metabolic adaptability, and govern the immune signature pattern in cells. The journey of ATG5 modulation from physiological developmental variation to pathological scenario brings out the translational impact of ATG5. On the one hand, ATG5 promotes exit from the pluripotent state by c-Myc degradation during differentiation of specific lineages (involved in neurogenesis, adipogenesis, and hematopoiesis), while on the other hand, it has a critical involvement in metabolic circuitry via rewiring autophagy through modulation of lipophagy, mitophagy, and acetyl-CoA epigenetics. Taken together, this work summarizes new findings that centrally place ATG5 as a driver that engineers a coordinated crosstalk between the metabolic state and cell fate decisions and immune responses. These insights position ATG5 as a critical and therapeutic target for developmental disorders, cancer, and immune-metabolic diseases.
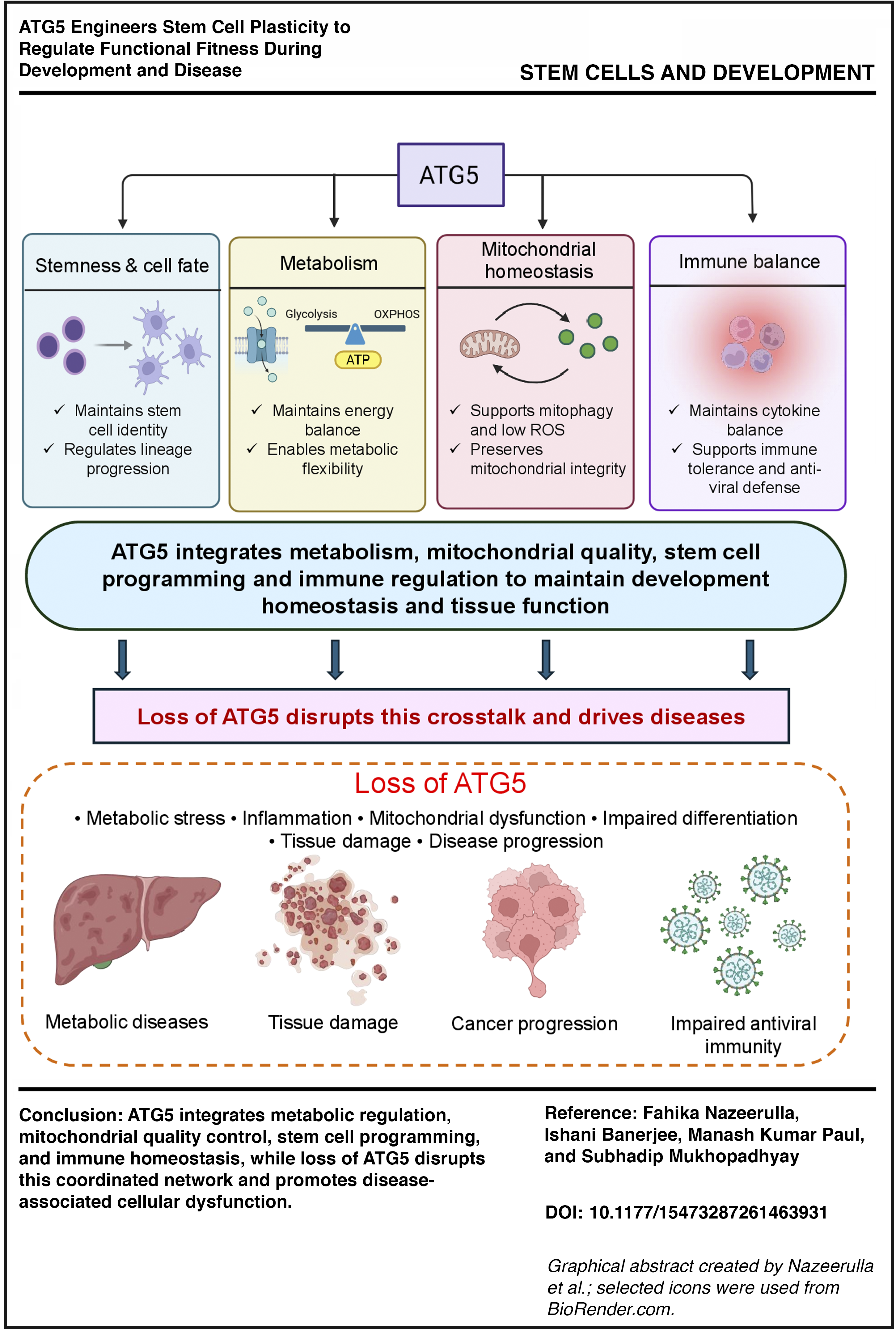
Introduction
Autophagy (Greek for “self-eating”) is a highly essential catabolic process that is constitutively involved in the management of cellular stress during a variety of crucial processes, including development, immunity, and cell death modulation, to name a few. Briefly, during the process of autophagy, a crescent-shaped initiating membrane starts elongating into a specialized double membrane structure called an autophagosome carrying nonessential cellular components, which later fuses with lysosomes to form autolysosomes. The extremely low pH of the autolysosomes helps in the breakdown of the autophagic cargo and releases free fatty acids and amino acids, which in turn help to circumvent the stressful situation.1,2 Autophagy drives the turnover of organelles, clearance of protein aggregates, and other macromolecules, thereby acting as a prime regulator of the cellular quality control center, which helps cells adapt to challenging stressful conditions.3,4 The steps involved in the autophagy pathway remain conserved across eukaryotes and are primarily governed by more than 40 ATG proteins. 5 In our latest study, we investigated how autophagy plays a major role as an architect of embryogenesis. Regardless of the functional diversity and different cellular origins of the organs like the lung, liver, heart, or brain, we found that ATG5 is the single most important and common autophagy protein that is crucial during organogenesis.
But till date, the detailed mechanism of ATG5-mediated remodeling of progenitor stem-like cells during physiological development or in the pathological context of cancer stem-like cells to regulate differentiation into different tissues with distinct metabolic needs mirroring an altered immune niche, remains unanswered and forms the central focus of this review. Autophagy, being an orchestrated procedure involving different proteins, remains difficult to target in the clinic, and unfortunately, till now, there is no single drug that has cleared the clinical trials and is available to patients solely targeting the autophagy pathway. Given the importance of ATG5, 6 understanding its role becomes imperative not only to decipher its critical signaling node affecting cellular plasticity, metabolism, and immunity but also to see it as an important drug discovery target.
Unless otherwise specified, in humans, ATG5 denotes the gene, while ATG5 refers to the protein. In the mouse system, Atg5 denotes the gene, whereas ATG5 refers to the protein. 7 During autophagosome formation, ATG5 is best characterized for its role a part of the ATG12-ATG5 conjugation system. 8 The restricted patterns observed for other ATG proteins, in contrast to the consistent requirement of ATG5 across multiple organ systems, show that it may occupy a central position within the autophagy network. 6 Evidence also suggests that ATG5 has functions beyond canonical macroautophagy (the ATG-dependent pathway that includes phagophore initiation, autophagosome formation, and lysosomal degradation), such as metabolic activity, cell fate decisions, and immune signaling.9–12 Together, these findings suggest that ATG5 may help coordinate several connected pathways that support normal development.
ATG5 Expression Across Development and Evolutionary Conservation Across Species
To examine this role more clearly, ATG5 expression was analyzed across developmental stages and tissue types using publicly available transcriptome data from the Human Protein Atlas and Temporal Expression during Development Database 2.0 (TEDD 2.0) (Fig. 1a).13,14 These datasets show that ATG5 is expressed across different developmental stages ranging from early stages of embryonic development to adulthood in both mice and humans, indicating that the function of ATG5 is not limited to a single time point in development but rather is maintained throughout life.
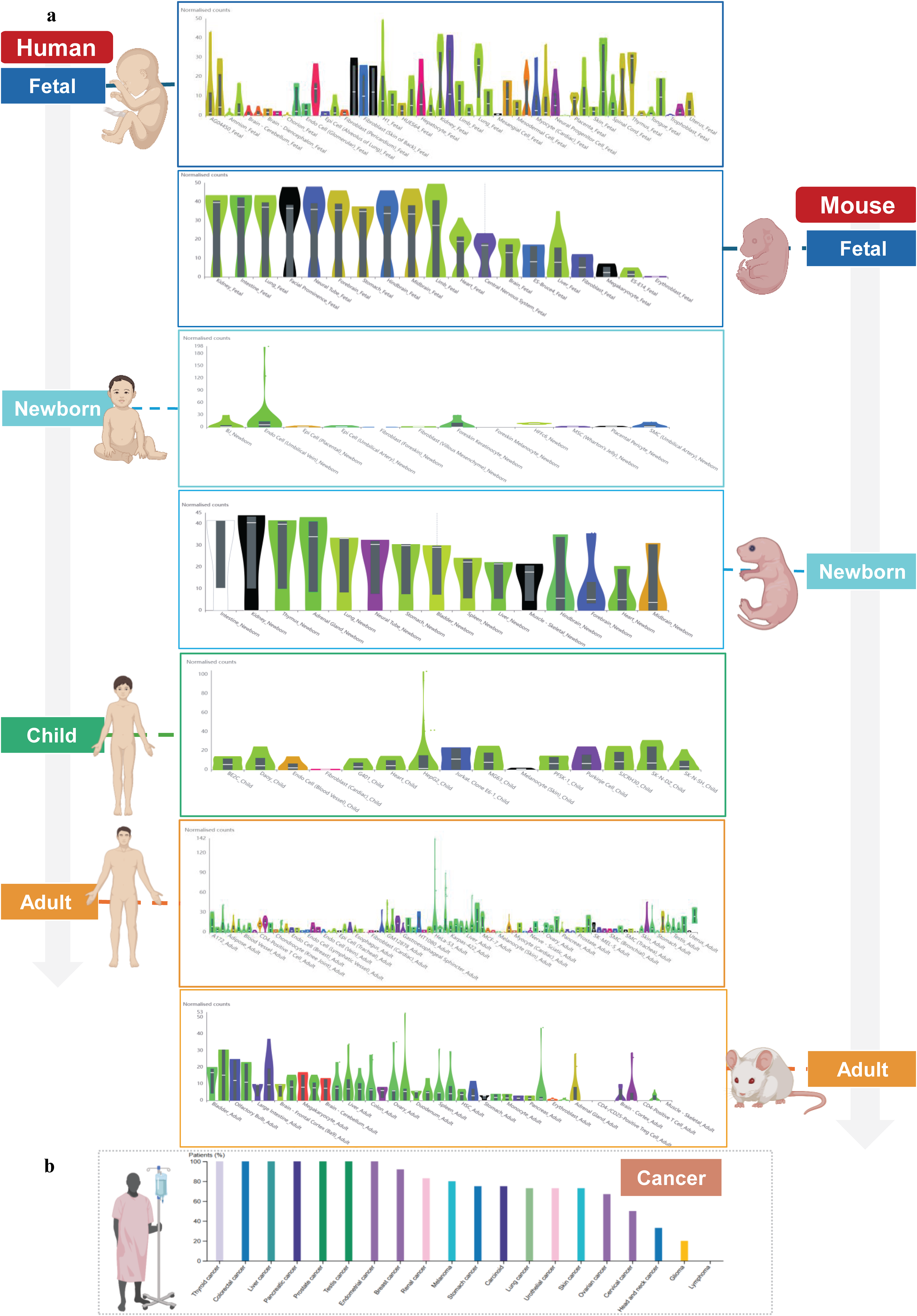
ATG5 expression was assessed across
Our analysis from Figure 1a shows that in both mice and humans, fetal tissues show the broadest ATG5 expression across multiple organ systems, compared to neonatal or postnatal tissues. In human fetal samples, expression is detectable in tissues including the brain, liver, heart, kidney, lung, and spinal cord. Similar to humans, widespread expression was also observed across mouse organs such as the kidney, intestine, lung, brain, and forebrain. In contrast, as development progresses into adults, ATG5 expression becomes more tissue-specific in neonatal, postnatal, and adult stages. From this, we can infer that ATG5 is broadly required during embryogenesis, which is consistent with the increased cellular remodeling demands of organogenesis. As tissue mature, ATG expression is restricted within specific tissues, reflecting the distinct needs of each organ. The conservation between mice and humans is not that ATG5 is expressed in the exact same tissues but the fact that both show a similar pattern of requirement for ATG5 across development, as mentioned above. This supports the idea that ATG5 has a conserved role in vertebrate development; however, ATG5′s exact tissue distribution may vary between species because each organism has different physiological demands.
Furthermore, as seen in Figure 1b, ATG5 levels are heterogeneous among tumor cells‚ suggesting that ATG5-dependent autophagy is regulated in a tumor- and context-specific manner. For instance, ATG5 may support tumor growth in certain contexts‚ such as via autophagy-dependent growth or via autophagy-dependent therapy resistance. It has been reported that ATG5-mediated autophagy has been associated with resistance to 5-fluorouracil in colorectal cancer‚ as ATG5 depletion through the homeobox containing 1 (HMBOX1) and HECT domain and ankyrin repeat-containing E3 ubiquitin protein ligase 1 (HACE1) pathway sensitizes cells to this chemotherapy drug. 15 Similarly, in prostate carcinoma‚ ATG5 upregulation downstream of Paired Box 5 (PAX5)–Isocitrate Dehydrogenase 1 Antisense RNA 1 (IDH1-AS1) stimulates autophagy-mediated tumor growth. 16 However, ATG5-dependent autophagy can also play a tumor-suppressive role, for example, spontaneous liver tumor formation in Atg5-null mice suggests the basal tumor inhibitory activity of ATG5-dependent autophagy in vivo. 17 Hence, the variable cancer-associated expression shown in Figure 1b should not be strictly interpreted as a pro- or antitumor signal but as context-dependent regulation of Atg5 across different cancers. 18
In addition to assessing the temporal pattern of human ATG5 expression, a multiple sequence alignment of ATG5 orthologs from seven vertebrate species (zebrafish, mouse, rat, human, bovine, chick, and pig) was performed using MultAlin to assess the evolutionary conservation of ATG5 (Fig. 2a). 19 A significant finding from this analysis was the very high degree of primary amino acid residue conservation. Approximately 74.1% of residues were found to be completely conserved (in red), while an additional 25.8% of residues were found to be partially conserved (in yellow); thus, overall, greater than 99% of residues were found to be either completely or partially conserved across the entire alignment. This conservation suggests that ATG5 cannot tolerate substantial sequence variation due to evolutionary selection favoring preservation of structural properties and interface features required for canonical and possibly noncanonical ATG5 functions. The phylogenetic tree generated based on ATG5 sequences corroborates the evolutionary conservation demonstrated by multiple sequence alignment, but also demonstrates the drawback of using a very highly conserved protein for a phylogenetic study (Fig. 2b). Zebrafish represented the most distantly related branch due to their early divergence from the amniotes, whereas chickens diverged from mammals due to vertebrate hierarchy. ATG5 sequences show such a high degree of similarity among mammals that no conclusions can be drawn regarding exact mammalian evolutionary relationships. The short branch lengths indicate that the ATG5 gene has not undergone much evolution due to its intolerance to mutation.

In summary, the above results collectively indicate that ATG5 expression is widely observed throughout the process of fetal organ formation, persists in postnatal and adult tissues, and is well-conserved in vertebrates. Nevertheless, the analysis of expression and conservation does not reveal the mechanism by which ATG5 functions on a molecular level, nor can it differentiate ATG5 availability from ATG5 activity or autophagic flux. Thus, the following discussion will begin with an overview of the role of ATG5 in the autophagy pathway.
ATG5 in Autophagy: Structure and Mechanistic Function
The involvement of ATG5 in influencing development-related and pathological programs begins with the fact that this gene product is part of the canonical macroautophagy pathway. One of the most important regulatory mechanisms of autophagosome biogenesis involves the ATG12 conjugation system where ATG5 is one of the key components as shown in Figure 3a. 20 Human ATG5 is an approximately 275-amino-acid-long protein consisting of two ubiquitin-like domains flanking a helical-rich region. 22 An important characteristic feature of ATG5 is its conjugation to ATG12 using an isopeptide bond formed between lysine residues, mediated by the E1-like enzyme ATG7 and the E2-like enzyme ATG10 (Fig. 3b). This ATG12-ATG5 conjugate forms a large complex with ATG16L1 in a stoichiometric ratio of 2:2:2 (Fig. 3c). This complex is recruited to the phagophore membrane and functions as a scaffold for other autophagy reactions.22–24 This means that ATG5 should not be considered just an adapter protein; rather, it should be viewed as an essential part of the molecular complex regulating the location of lipidation of LC3/GABARAP.
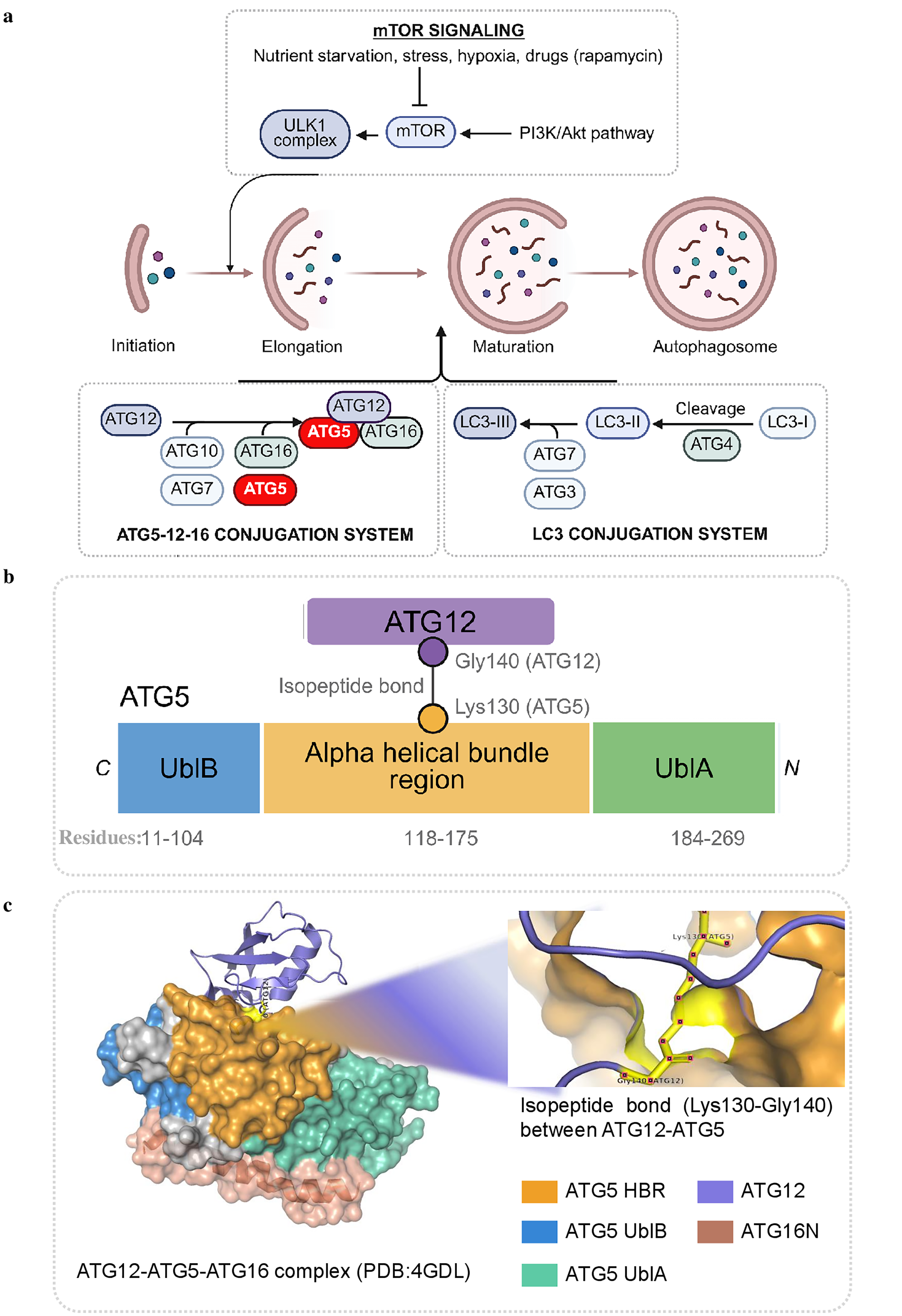
Structural and mechanistic role of ATG5 in autophagy.
Regarding its functions, the ATG12-ATG5-ATG16L1 complex has an E3 ligase-like activity within the ATG8/LC3 conjugation cascade involved in transferring LC3/GABARAP proteins from the cytosol to phosphatidylethanolamine (PE) on the autophagosomal membrane.22,25 This lipidation step is crucial since it provides microtubule-associated protein 1 A/1B-light chain 3 (LC3) with the ability to associate with the autophagic membrane, expand the membrane surface, recruit cargo, and mature vesicles. 23 Moreover, ATG5 is involved in later stages of the autophagic pathway through interaction with tectonin beta-propeller repeat-containing protein 1 (TECPR1) and regulates autophagosome-lysosome fusion. 26 Therefore, ATG5 is engaged not only in the early stages of membrane expansion but also in the terminal stages of autophagic flux. It can be concluded, based on genetic evidence and functional analyses, that ATG5 is required for constitutive expression of autophagy.22,26,27 The absence of ATG5 causes complete dysfunction and failure of the autophagic process, indicating its critical role among the core components necessary for autophagosome production. Together, ATG5 is confirmed as an important element of the canonical autophagy pathway. Nevertheless, the importance of ATG5 for development cannot be explained by its participation in autophagy alone. The role of stem and progenitor cells in this context becomes especially obvious because the decision to undergo differentiation is associated with extensive cell reorganization. This aspect will be described in more detail in the next paragraph.
ATG5 as a Regulator of Stem Cell Fate and Lineage Progression
The importance of ATG5 has been further stressed by its specific role in stem and progenitor cells‚ where many differentiation transitions during embryogenesis‚ tissue homeostasis, and repair take place, making them sensitive to changes in proteostasis‚ mitochondrial quality control, and inflammatory tone. The context-dependent roles of ATG5 have been shown in studies of embryonic stem cells‚ neural stem cells‚ hematopoietic stem cells‚ epithelial stem cells, and cancer stem cells.
ATG5 as a regulator of pluripotency exit
The strongest evidence for a role for ATG5 in stem cell fate determination is in pluripotent stem cells‚ where ATG5 promotes exit from pluripotency and acquisition of differentiation potential. In mouse ESCs, ATG5 supports a subdued inflammatory state that is necessary for differentiation by means of particular mechanisms; it attracts F-box and WD-40 domain protein 7 (FBXW7) to initiate beta-transducing repeat containing E3 ubiquitin protein ligase 1 (β-TrCP1) degradation, thus downregulating nuclear factor κB (NF-κB) signaling. Deficiency in ATG5 leads to inflammatory gene activation and differentiation problems.28,29 In parallel, ATG5 directly suppresses cellular myelocytomatosis oncogene (c-Myc)-associated growth signals linked to stemness by physically interacting with c-Myc and recruiting FBXW7, resulting in c-Myc degradation by the proteasome, facilitating exit from pluripotency. 9 Together, these studies suggest that ATG5 promotes exit from pluripotency by selectively suppressing molecular programs that mediate pluripotency maintenance or inhibit differentiation‚ rather than by broadly stimulating cellular degradation.
ATG5 in lineage differentiation, maturation, and survival
Moreover, ATG5 is often required not only for differentiation but also for the remodeling that allows newly specified cells to mature and survive. ATG5 facilitates neuronal differentiation during embryonic cortical development, but limits excessive proliferation by altering β-catenin signaling. 30 Atg5-null olfactory bulb stem/progenitor cells fail to differentiate into neurons, suggesting ATG5-dependent autophagy is required for neurogenesis. 31 Remarkably, its function is not limited to lineage determination. Although the differentiation is not affected, ATG5 is strongly needed post differentiation. ATG5 helps maintain the survival and maturation of the neural progenitor-derived cells by blocking BCL-2-associated X protein (BAX)-dependent apoptosis. 32 ATG5 also regulates astrocyte differentiation through upregulation of suppressor of cytokine signaling 2 (SOCS2) degradation, leading to activation of Janus kinase 2-signal transducer and activator of transcription 3 signaling gliogenic pathway‚ which causes astrocyte-specific gene transcription and glial fibrillary acidic protein (GFAP)-positive astrocyte formation. 33
The dependency on ATG5 is not limited to neural tissues. For example, during adipogenesis, ATG5 is not necessary for the early differentiation of adipocytes but is essential for late-stage cytoplasmic remodeling and mature lipid droplet formation. 34 In B cells‚ ATG5 promotes late activation and plasma cell differentiation via induction of syndecan-1/CD138 expression and plasma cell transcriptional regulators such as PR domain zinc finger protein 1 and X-box binding protein 1, which are critical for efficient immunoglobulin secretion. 35 Taken together, these studies indicate that ATG5 is a factor that not only determines lineage specification but also ensures proper maturation of differentiated cells.
ATG5 in adult stem cell maintenance, self-renewal, and regeneration
Within adult stem cell compartments‚ ATG5 protects regenerative capacity against the loss of self-renewal caused by cellular stress. For example, autophagy mediated by ATG5 in salivary gland stem cells increases during the transition of cells from quiescent to active self-renewal. Its deletion leads to the gradual loss of self-renewal capacity of the cells without a corresponding increase in cell death. 36 Similarly, ATG5 is involved in the support of leucine-rich repeat (LRR)-containing G protein-coupled receptor 5 intestinal stem cells maintenance and their recovery after stress through the downregulation of reactive oxygen species (ROS) buildup and endoplasmic reticulum (ER) stress. 37 In adult epidermal, dermal, and hematopoietic stem cells, ATG sustains self-renewal, differentiation potential, and resistance to cytotoxic stress. 38 Thus, in adult stem cells, ATG5 mainly functions as a fitness-preserving factor that enables regenerative programs to proceed without excessive oxidative, proteotoxic, or ER stress.
ATG5 in hematopoietic stem cell development and metabolic adaptation
ATG5 links developmental progression with metabolic fitness in hematopoiesis. For example, endothelial-specific Atg5 deletion blocks endothelial-to-hematopoietic transition, and pre-hematopoietic stem cells (HSCs) fail to mature due to altered nucleolin signaling, runt-related transcription factor 1/growth Factor Independence 1 regulation, mitochondrial metabolism, and protein homeostasis. 39 At the same time, ATG5 protects neonatal HSCs from mitochondrial oxidative stress, and its function is long lasting in the hematopoietic system independently of p62. 40 In hematopoietic stem and progenitor cells, dysregulated mitochondrial quality control, elevated ROS, glycolytic metabolism, and increased proliferation are linked to reduced ATG5-dependent autophagy, suggesting that loss of ATG5 may create an environment that favors leukemic transformation. 41
Niche-dependent and pathological stem cell regulation by ATG5
ATG5 can also indirectly affect stem cell function via its role in the niche and in pathological states by cancer stem cell programming. In the Drosophila ovary, ATG5 is essential for bone morphogenetic protein-phosphorylated Mad signaling in cap cells, which is required to preserve the stem cell niche integrity, thus playing a role in the maintenance of GSCs within the Drosophila ovary. 42 Interestingly, in Alzheimer’s disease, microglial ATG5 is thought to help maintain hippocampal neural stem cells (NSCs) and neurogenesis in a disease context-dependent manner. 43 Yet, cancer stem cells appear to exploit ATG5 to maintain their pathological stemness. For example, ovarian cancer stem cells need ATG5-forkhead box A2 signaling for their self-renewal and chemoresistance, 44 whereas liver cancer stem cells depend on a tissue differentiation-inducing nonprotein coding RNA–polypyrimidine tract binding protein 1–ATG5 axis for their self-renewal and tumorigenesis. 45
Besides the literature-derived data, a complementary database-data driven analysis was performed to systematically identify potential associations of ATG5 with stem cell regulatory proteins. The human ATG5 interactors were obtained from BioGRID and compared with a curated list of well-known stemness and differentiation markers (Supplementary Tables S1, S2, and S3).46–55 This analysis identified three proteins: MYC, STAT3, and TUBB3, related to stem cell regulation that interact with ATG5. After conducting further evaluations through a targeted literature review, we found that ATG5 affects those candidates in different manners. For instance, MYC, which was identified as an interactor, is mainly regulated by ATG5 via autophagy-independent mechanisms. 9 Therefore, MYC is not shown in Figure 4, which only focuses on autophagy-dependent functions of ATG5. The role of MYC is however presented in the “ATG5 as a Regulator of Pluripotency Exit” section. Conversely, molecular studies reveal an ATG5–STAT3 interaction that causes SOCS2 degradation and leads to transcriptional activation of astrocyte differentiation genes. 33 Moreover, TUBB3 expression, which serves as a neuronal differentiation marker, is in accord with ATG5 activity, even though the direct mechanistic connection is still unclear. 30
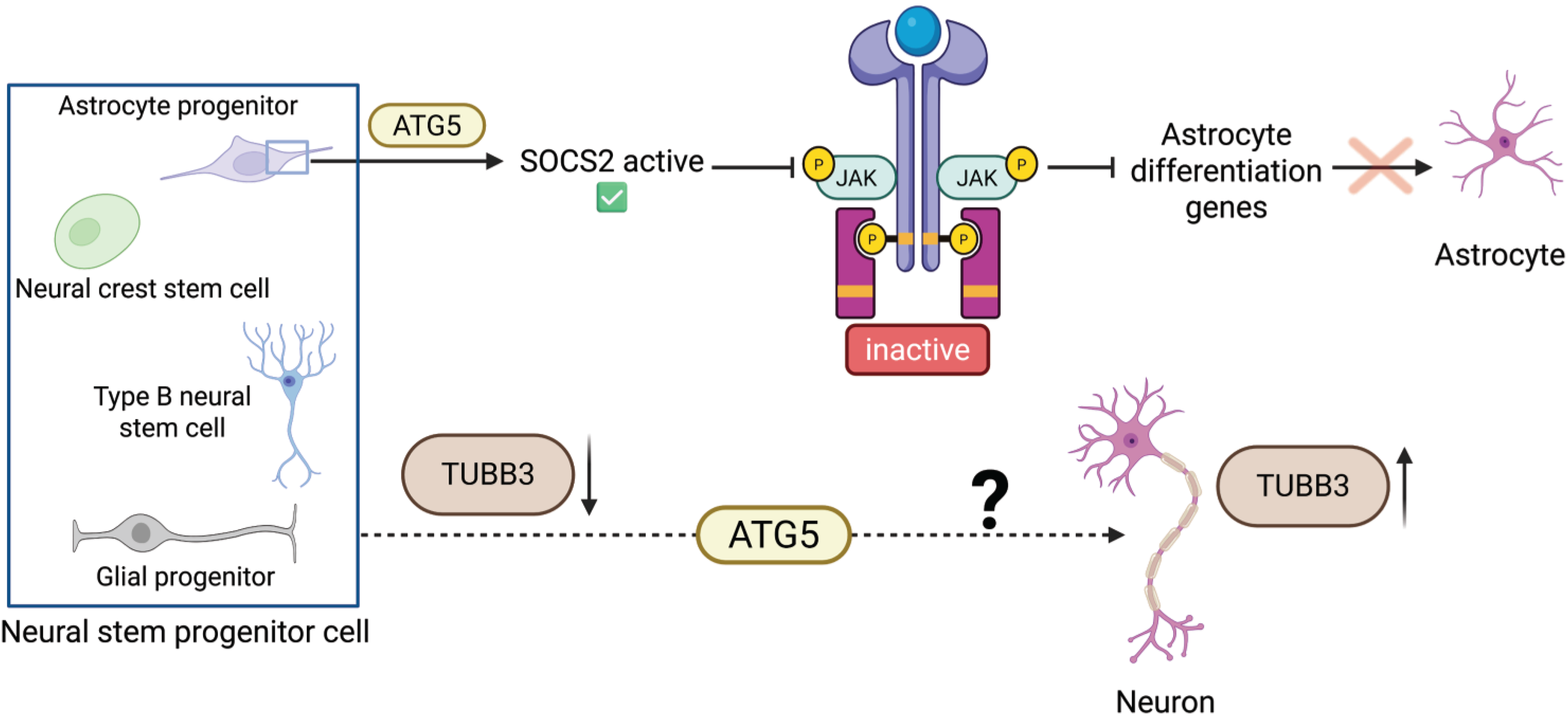
ATG5-associated differentiation pathways identified from BioGRID-guided analysis. The schematic highlights two ATG5-associated candidates linked to neural lineage differentiation. ATG5 promotes SOCS2 degradation in astrocyte progenitors, enabling JAK2–STAT3 activation and astrocyte differentiation gene expression. TUBB3 is shown as a neuronal maturation marker associated with ATG5-dependent neurogenic progression, although the direct mechanistic relationship remains unclear. Images created using BioRender. JAK2–STAT3, Janus kinase 2-signal transducer and activator of transcription 3; SOCS2, suppressor of cytokine signaling 2.
Thus, the findings presented in this section indicate that ATG5 plays an important role in cell-fate plasticity by ensuring stem cells can maintain, exit, or redirect cell-fate pathways depending on developmentally and disease-related needs. As several of these changes necessitate modifications in mitochondrial function, lipid modulation, and metabolic activity, the following section considers how ATG5 controls cellular metabolic reprogramming in cells.
ATG5 as a Central Regulator of Cellular Metabolic Reprogramming
ATG5′s ability to regulate stem cell fate is highly associated with its role in regulating cellular metabolism. Metabolic fitness is enabled through ATG5′s ability to regulate lipid mobilization, mitochondrial quality control, glucose metabolism, and availability of metabolic substrates. However, rather than functioning simply as a maintenance factor, the regulation by ATG5 plays an essential part in transcriptional programs and metabolic plasticity under stressful situations or diseases.
ATG5-dependent lipophagy fuels lipid remodeling and fatty acid oxidation
One of the major roles of ATG5 is in lipid metabolism, where it particularly acts through lipophagy. In the context of adipocyte differentiation, ATG5 plays a critical role during the late stage, as the absence of Atg5 results in inhibition of cytoplasmic remodeling and lipidation of ATG8/LC3 protein, which limits mature lipid droplet formation in the mouse embryonic fibroblast adipogenesis model and also adipocyte differentiation in vivo. 34 In hepatocytes, ATG5-mediated lipid removal supplies fatty acids for beta-oxidation and helps prevent triglyceride accumulation. 56 During nutrient stress, ATG5 also supports energy balance and lipid utilization by promoting fatty acid mobilization. 57 Furthermore, ATG5 also acts at the transcriptional level. ATG5 facilitates nuclear receptor co-repressor 1 degradation, which leads to activation of peroxisome proliferator-activated receptor alpha-driven metabolic gene expression, thereby promoting beta-oxidation and ketogenesis. 58 ATG5 thus regulates lipid metabolism in connection to energy generation and lineage-specific cell remodeling.
ATG5 preserves mitochondrial quality and metabolic resilience
In addition, ATG5 has a role in mitochondrial functioning and energy balance. It enables clearance of faulty mitochondria by mitophagy and ensures metabolic balance and insulin responsiveness. 59 In Atg5 transgenic mice, increased ATG5 expression results in increased autophagic flux and a lean phenotype, with enhanced glucose and insulin tolerance, low levels of triglycerides and leptin, maintained oxidative-reduction balance in the liver, and increased mitochondrial oxygen consumption in mouse embryonic fibroblasts, thus showing the impact of ATG5 on metabolic homeostasis. 60 On the contrary, beta-oxidation and ATP production are compromised in ATG5 deficiency conditions, thereby increasing sensitivity to cellular metabolic stress. 61 These studies indicate the involvement of ATG5 in regulation of mitochondrial function and metabolic fitness.
ATG5 links lipid-derived acetyl-CoA to lineage gene regulation
In lymphatic endothelial cells, ATG5 has a role in linking lipid metabolism with epigenetic regulation of lymphatic gene expression. ATG5-dependent lipophagy results in the delivery of fatty acids from lipid droplets to mitochondria, where fatty acids undergo fatty acid oxidation to form acetyl-CoA. This molecule is then used in the acetylation of histone H3K9 in E1A binding protein p300-mediated acetylation of prospero homeobox 1 (PROX1)-regulated lymphatic genes such as carnitine palmitoyltransferase I. Loss of Atg5 results in defective lipid droplet degradation and decreased acetyl-CoA and H3K9 acetylation, and ultimately impair PROX1-driven lymphatic gene regulation and lymphangiogenesis. This effect can be reversed through acetate supplementation.62,63
ATG5 rewires glucose handling in cell- and disease-specific contexts
Additionally, ATG5 has been reported to regulate glucose metabolism. In neurons, it regulates glycolysis flux by mediating the degradation of glucose transporter type 2, with its deficiency giving rise to excess uptake of glucose and hyper glycolysis. 10 In pancreatic β-cells, ATG5 is essential for glucose-stimulated insulin secretion, further demonstrating that autophagy is involved in glucose-sensing in metabolic regulation. 64 However, glucolipotoxicity, a phenomena related to elevated fatty acid and glucose levels, also induces excess ATG5 activity, leading to β-cell dysfunction and apoptosis, indicating the context-specific differences in the role of ATG5. 65
Under pathological conditions, ATG5 promotes metabolic reprogramming of immune cells and cancers. In microglia, ATG5 maintains glucose uptake and glycolysis to provide neuroprotection in Alzheimer’s disease. 66 In nonalcoholic fatty liver disease (NAFLD)-associated hepatocellular carcinoma, ATG5 supports tumor development through lipophagy-dependent lipid utilization. 67 The biological consequences of ATG5-dependent metabolism are thus determined by whether the adaptive metabolic responses restore physiological tissue activity or facilitate disease-associated survival. The metabolic activities associated with ATG5-dependent metabolism do not exist in a vacuum. By regulating mitochondrial fitness, lipid metabolism, glucose metabolism, and metabolic availability, ATG5 also regulates the metabolic environment that is necessary for the activation of immune cells, inflammatory tone, and antigen presenting capacity. Therefore, we next examine Atg5 as a regulator of immune balance.
ATG5-Dependent Immune Calibration: Activation, Restraint, and Tolerance
In both innate and adaptive immunity compartments, ATG5 acts as a calibrator of immune balance rather than a mere immune activator or suppressor. ATG5 can support the survival of immune cells, suppress inflammatory signals, or promote antigen presentation or limit immunopathology based on cell type and stimulus.
ATG5 maintains adaptive immune cell fitness and fate control
ATG5 is critical for lymphocyte homeostasis in the adaptive immune system. ATG5 restricts pathogenic T helper 17 (Th17) differentiation in cluster of differentiation 4 (CD4+) thymus-derived cells (T cells) by inhibiting reprogramming of glycolysis and production of interleukin 17A (IL-17A) and granulocyte-macrophage colony-stimulating factor (GM-CSF), thereby limiting inflammation and tissue fibrosis. 11 In contrast, as a negative regulator of effector functions in CD8+ T cells, ATG5 ablation improves cytotoxicity, cytokine production, and tumor clearance by metabolic and epigenetic reprogramming. 68 In addition to its functional regulation, ATG5 is also needed for T cell survival and clonal expansion, as loss of ATG5 caused apoptosis and inhibition of expansion, despite activation. 69 In bone marrow-derived cells, ATG5 is needed at early developmental checkpoints and for cell survival, most importantly for the maintenance of B1a cell populations. 70 In summary, the data presented above suggest that ATG5 regulates adaptive immunity through the maintenance of the fitness of lymphocytes and their differentiation into proper effector cells depending on cell type and context.
ATG5 restrains innate inflammatory signaling
ATG5 also plays a role in innate immunity by negatively regulating inflammation. Disruption of ATG5 in myeloid cells improves NF-κB/mitogen activated protein kinase (MAPK) signaling and IL-23 production, which results in systemic inflammatory responses and metabolism dysregulation. 71 Furthermore, in macrophages, ATG5 acts to balance between M1/M2 polarization; its absence favors M1 phenotypes with enhanced production of tumor necrosis factor (TNF) and IL-6. 72 Moreover, ATG5 is necessary to restrain immunopathology during infection, by restricting the overproduction of cytokines and the recruitment of neutrophils to sites of infection without affecting pathogen elimination. 73 In the innate immunity, ATG5/ATG12 complex suppresses antiviral signaling by interacting with retinoic acid-inducible gene I and mitochondrial antiviral signaling protein, thereby inhibiting the production of type I interferon. 74
In relation to granulocytes, ATG5 regulates their differentiation process. Deficiency of ATG5 causes increased proliferation of neutrophils, whereas eosinophilia development is impaired. 75 Finally, regarding the role of ATG5 in macrophages, ATG5-driven autophagy plays an important part in antitumor processes via the maintenance of antigen presentation and inhibition of programmed cell death-ligand 1-mediated immune suppression in macrophages. Loss of ATG5 leads to M2 like polarization and tumors development. 76 As seen from the results discussed above, ATG5 regulates innate immunity by controlling multiple aspects of the processes in question, such as cytokine signaling, antiviral responses, and myeloid cell lineage output.
ATG5 shapes antigen presentation and T cell priming
ATG5 is also required for antigen processing and presentation by dendritic cells, where it regulates phagosome–lysosome fusion during major histocompatibility complex (MHC) class II presentation of antigens and CD4+ T cells priming. 77 ATG5-dependent LC3-associated phagocytosis enhances presentation of microbial and self-antigens via MHC class II, supporting host immunity but also promoting autoimmune T cell activation. 78 ATG5 also regulates dendritic cells immunometabolism; its loss increases glycolysis, cytokine release, and MHC class I expression, thereby enhancing CD8+ T cell activation. 79 The findings presented above demonstrate that the effects of ATG5 include antigen presentation support/restriction depending on antigen processing pathway, immune stimulus, and involved T cell populations.
ATG5 dysregulation in autoimmunity and inflammatory disease
ATG5 dysregulation is implicated in autoinflammatory and autoimmune disorders. For example, ATG5-dependent LC3-associated phagocytosis is important for clearance of dying cells, and its deletion leads to lupus-like autoimmunity. ATG5 variants also impact NF-κB signaling and type I interferon signaling pathways, increasing disease susceptibility. 26
Taken together, ATG5 regulates immune balance through the maintenance of lymphocyte fitness, suppression of excessive innate immunity activation, modulation of antigen presentation, and promotion of immune tolerance. To determine whether these effects operate independently or are interconnected with developmental and metabolic processes, an integrated proteomic analysis was performed using Atg5-null systems as discussed in the next section. Table 1 summarizes the ATG5 activity related to stemness, metabolism, and immunity with respect to cell-type and -context.
Context-Dependent Roles of ATG5 in Stem Cell Fate, Metabolism, and Immune Regulation
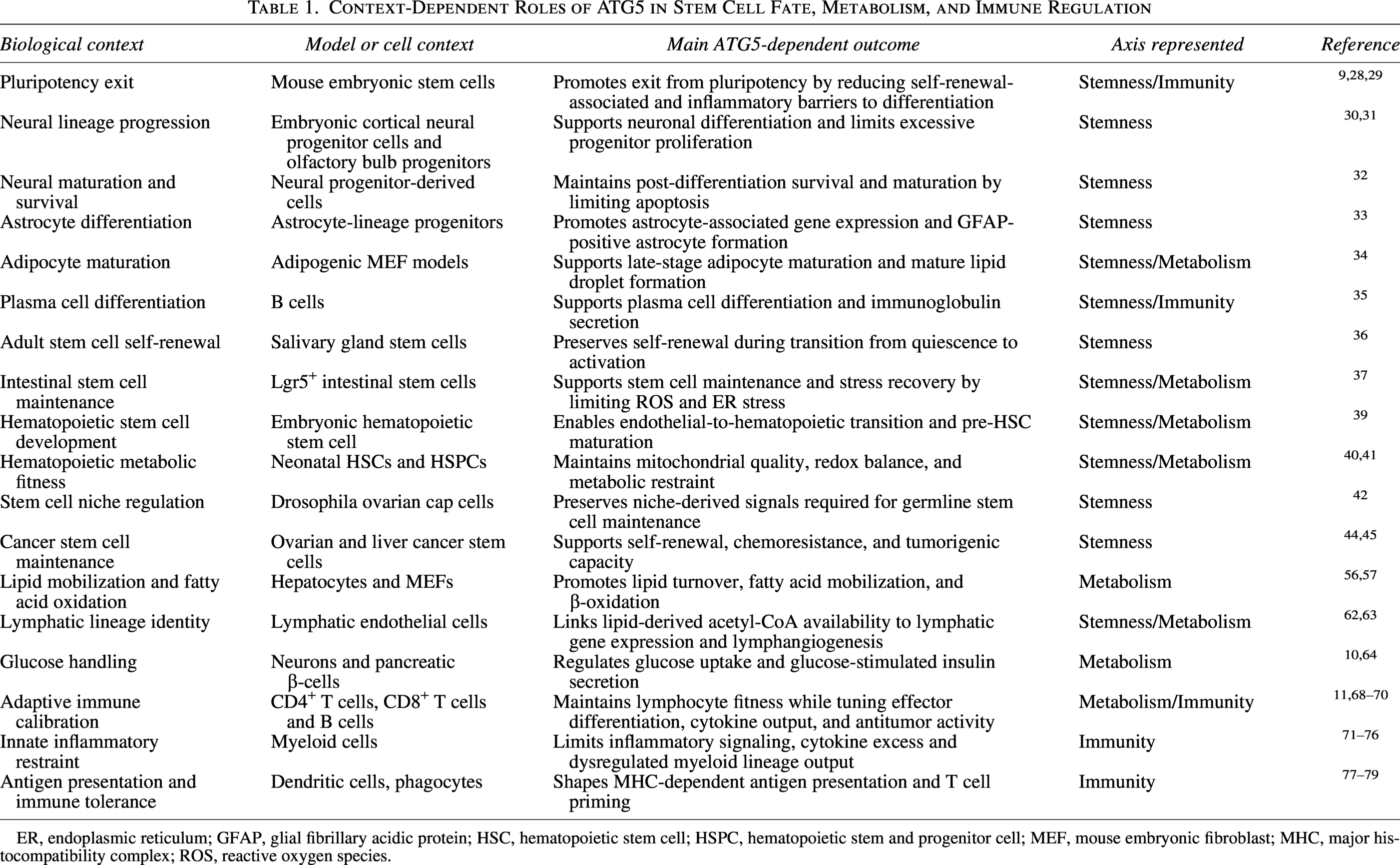
ER, endoplasmic reticulum; GFAP, glial fibrillary acidic protein; HSC, hematopoietic stem cell; HSPC, hematopoietic stem and progenitor cell; MEF, mouse embryonic fibroblast; MHC, major histocompatibility complex; ROS, reactive oxygen species.
Atg5-KO Proteomics Reveals Coordinated Dysregulation Across Stemness, Immune, and Metabolic Programs
To provide a broader view of biological processes affected by Atg5 loss, GeneCodis was used to perform GO enrichment analysis on a publicly available proteomics dataset from Atg5 knockout (KO) cells.80,81 GO terms of both upregulated and downregulated proteins were analyzed, and biological processes related to stemness/differentiation/development, immune regulation, and metabolism were identified. Proteins in these three categories were then extracted and analyzed using volcano plots with fold-change cutoffs of −1.5 and 1.5, with significance set at −log10 P value >2 (Fig. 5).
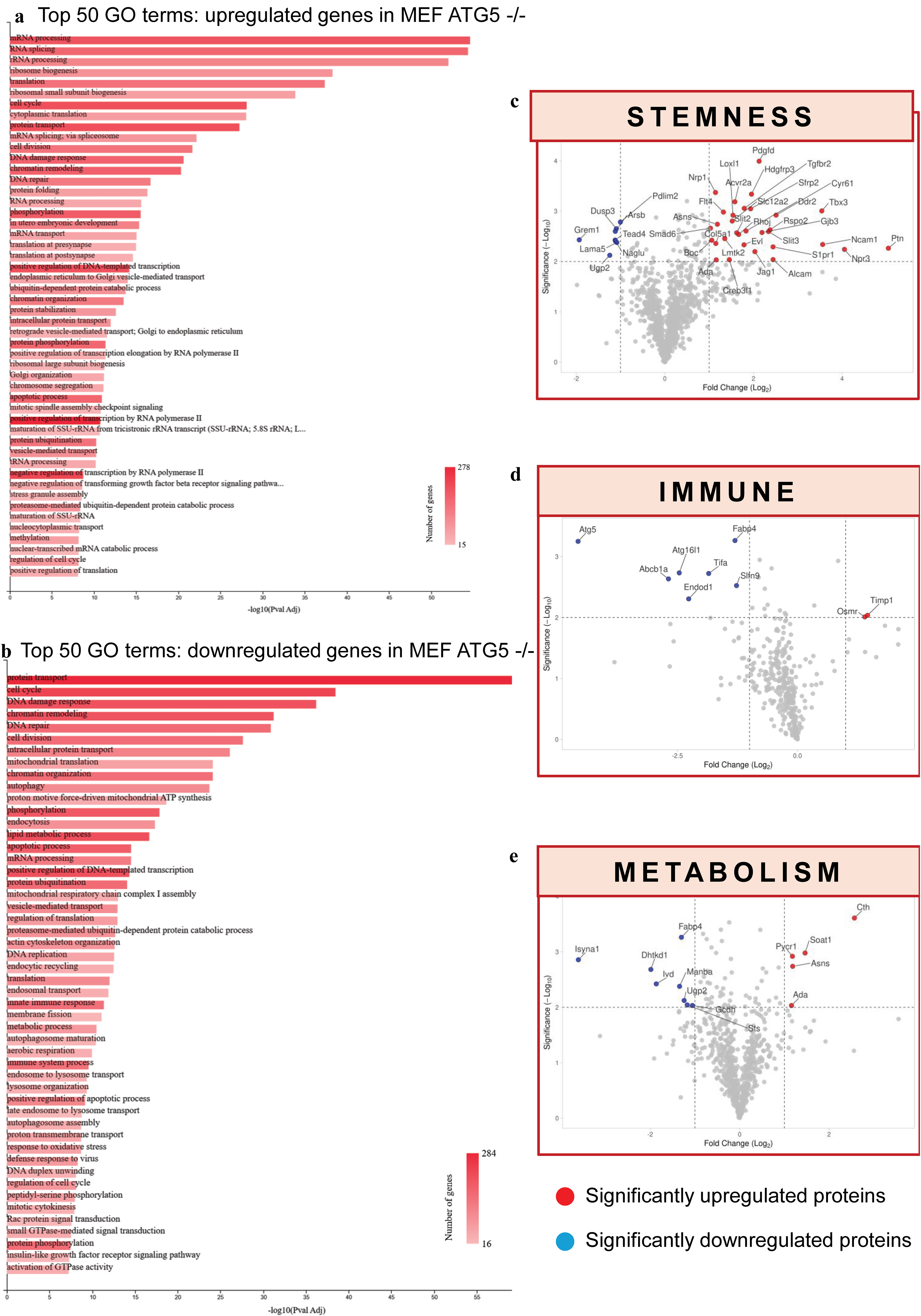
Proteomic and Gene Ontology (GO) enrichment analysis of Atg5 knockout reveals coordinated dysregulation of developmental, immune, and metabolic pathways.
The data show that Atg5 deficiency affects several major biological processes at the same time rather than a single pathway. As seen in Figure 5a, the top 50 GO terms of the upregulated proteins showed increased messenger RNA (mRNA) processing, ribosome biogenesis, cell cycle activity, and DNA damage response, consistent with release of proliferative programs following loss of ATG5-mediated c-Myc degradation. 9 In contrast, downregulated proteins (Fig. 5b) were linked to autophagy, mitochondrial respiratory chain, innate immune response, and lipid metabolism. This supports autophagy failure and matches the roles of ATG5 in mitophagy, lipophagy, and immune regulation discussed in Sections 7 and 8.
The three volcano plots (Fig. 5c–e) showed proteins linked to stemness were mainly upregulated after Atg5 loss such as the Notch ligand Jagged1 and neural stem cell mitogen pleiotrophin. Together, these changes suggest that cells failed to leave the stem-like state upon ATG5 loss, which fits with the reported role of Atg5 in pluripotency exit and lineage progression.29,33,82,83 In contrast, immune and metabolic proteins were mostly downregulated, suggesting weakened immune and metabolic functions.10,69,77 In the immune panel (Fig. 5d), reduced ATG16L1 suggests disruption of the ATG12-ATG5-ATG16L1. Increased fatty acid-binding protein 4 (FABP4) suggests a shift toward pro-inflammatory activation, matching M1 macrophage skewing described in the “ATG5 Restrains Innate Inflammatory Signaling” section.72,84 In the metabolic panel, lower levels of the mitochondrial enzymes dehydrogenase E1 and transketolase domain-containing 1 support failure of mitochondrial function and ATP production. 85 This aligns with the study presented in the “ATG5 Preserves Mitochondrial Quality and Metabolic Resilience” section, which describes loss of Atg5 impairs ATP production. 61
Interestingly, FABP4 appeared in both immune and metabolic panels. UDP-glucose pyrophosphorylase 2 appeared in both the metabolic and stemness panels. These overlaps give direct molecular support for the link between immune and metabolic changes, as well as the link between metabolism and developmental state. Hence, this provides a foundation for the next section, which discusses examples where ATG5 connects cell fate, metabolism, and immune regulation within the same biological system.
ATG5 at the Interface of Cell Fate, Metabolism, and Immunity: Coordinating Developmental Programs
Emerging evidence shows that ATG5-dependent autophagy acts as a hub connecting metabolic reprogramming with regulation of cellular identity, stemness, and immune responsiveness (Fig. 6).
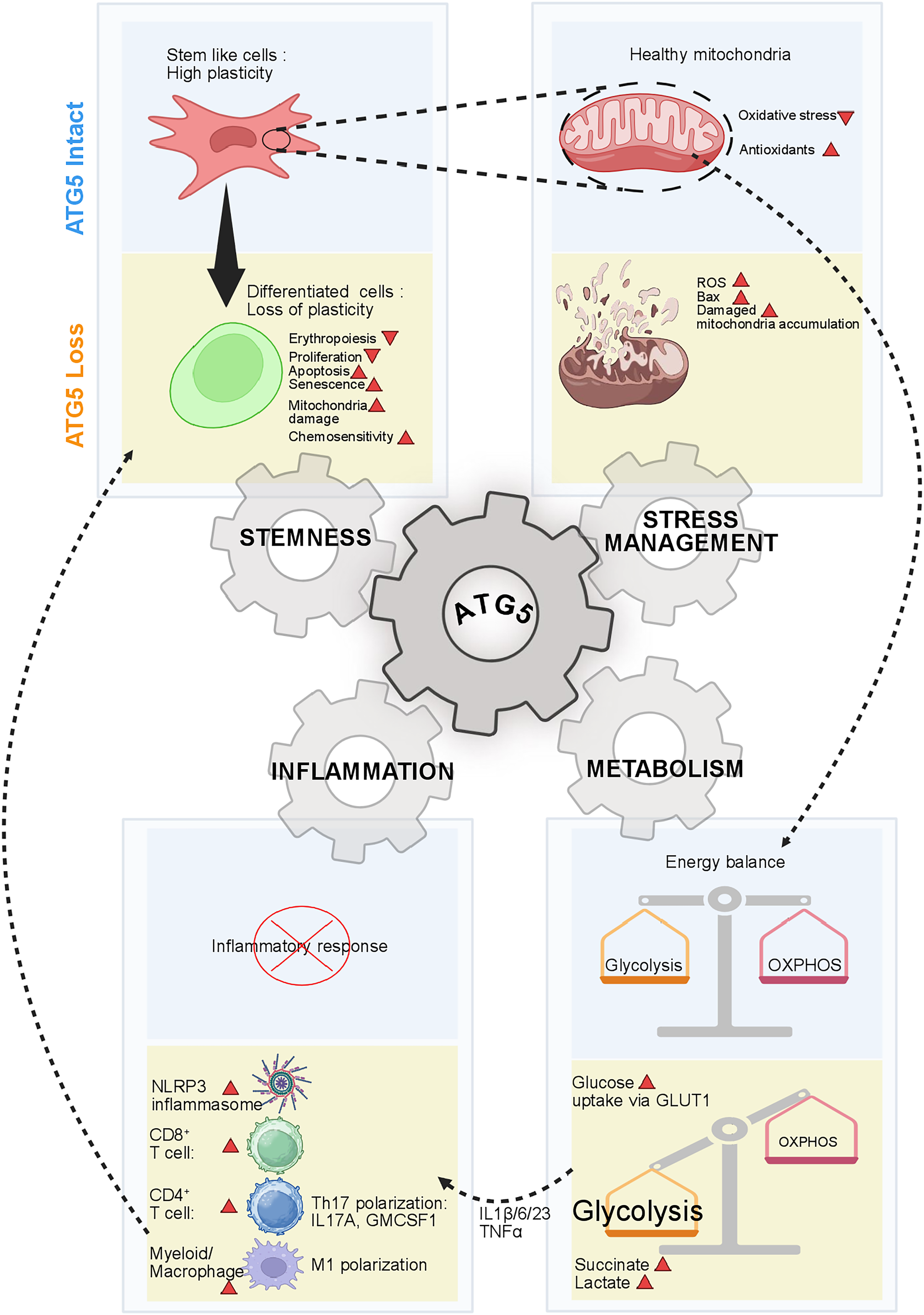
ATG5 supports stem cell maintenance, mitochondrial integrity, balanced glycolysis/oxidative phosphorylation metabolism, and suppression of inflammatory activation. Loss of ATG5 promotes differentiation-associated defects, mitochondrial damage, oxidative stress, a glycolytic shift, inflammasome activation, and pro-inflammatory immune polarization, suggesting that ATG5 coordinates multiple cellular programs that are essential for developmental homeostasis. Image created in BioRender. Mukhopadhyay, S. (2026) https://BioRender.com/pjmrxd2.
Atg5 suppression in lymphatic endothelial cells results in lipid droplet accumulation and elevated intracellular triglycerides through defective lipophagy, which leads to decreased fatty acid oxidation and mitochondrial dysfunction. This results in decreased acetyl-CoA/CoA levels, which in turn reduces PROX1-dependent lymphatic genes. These data demonstrate that the loss of PROX1-dependent transcriptional programs is accompanied by altered cell viability, implying a transition from a fully differentiated phenotype to a more plastic and possibly dysfunctional phenotype. Atg5 knockout cells are also defective in the response to VEGF-C (Vascular endothelial growth factor C) and could have more widespread physiological implications in immune cell trafficking and tissue repair. 62
In immune cells like CD4+ T cells autophagy driven by ATG5 is one of the key determinants of immunometabolic programming. In both human and mouse models, the reduction of ATG5 levels resulted in a disruption in autophagy and metabolic reprogramming, which involves an increase in the expression of genes responsible for the glycolysis pathway (Hexokinase 1, Hexokinase 2, Pyruvate Kinase M, Glucose-6-Phosphate Isomerase, Aldolase A, Phosphofructokinase, Phosphoglycerate kinase 1, Glyceraldehyde 3 phosphate dehydrogenase, Enolase 1 and Hypoxia-Inducible Factor 1-alpha). The reprogrammed metabolism is associated with an alteration in its cellular identity where CD4+ T cells adopt the pathogenic Th17 (IL-17A+ IFN-γ+) phenotype. Notably, there is an elevation in the production of IL-17A and GM-CSF, accompanied by the expression of Th17-specific transcription factors. These metabolic reprogrammed T cells coordinate a multicellular inflammatory network through cytokine signaling; as a result, these T cells enhance the activity of type 3 inflammation pathways. This highlights the importance of ATG5-driven metabolism in regulating T cell function. 11
In many CD8+ T cells, Atg5 deficiency promotes production of pro-inflammatory cytokines along with enhanced glycolytic metabolism. The loss of Atg5 impairs mitophagy, leading to mitochondrial dysfunction and ROS accumulation, which increases oxidative stress in immune cells, and this together with a shift toward glycolytic metabolism drives NLRP3 inflammasome activation, resulting in the release of IL-1β and IL-18. 86
Another study emphasizes the importance of Atg5-dependent autophagy for the maintenance of metabolic and immune balance of liver CD11c+ myeloid cells. Atg5 deletion results in systemic metabolic disturbances along with hepatic steatosis and elevated triglycerides, characteristic of NAFLD. Atg5 deficiency also drives a pro-inflammatory immune shift by enhancing IL-23 production in CD11c+ cells, mediated through p38 MAPK and NF-κB signaling pathways. The blockade of the IL-23-driven inflammatory axis restores metabolic homeostasis and reduces liver inflammation. Atg5 loss promotes the accumulation of CD11c+ dendritic cells and inflammatory macrophage populations, further amplifying hepatic inflammation. 71
The suppression of ATG5 in aged macrophages actually impairs autophagic flux, which directly drives the differentiation of macrophages toward an M1 pro-inflammatory phenotype with upregulation of inducible nitric oxide synthase, TNF-α, IL-1β, IL-6, monocyte chemoattractant protein-1, and decreased expression of M2 markers (Arginase-1 and Mannose Receptor C-Type 1). Mechanistically, this is supported by increased STAT1 activation and decreased STAT6 signaling which leads to a consolidation of the inflammatory state. Functionally, ATG5-deficient macrophages are characterized by an increased senescence-associated secretory phenotype characterized by increased levels of TNF-α, IL-1β, and IL-6, which further promote neutrophil recruitment and hepatocyte injury, thus aggravating acute liver injury. These effects are reversed by ATG5 restoration, which restores autophagy and macrophage polarization, thus identifying ATG5 as an important regulator of the crosstalk between the immune and differentiation systems. 87
Atg5 deficiency in microglia disrupts the acquisition of disease-associated microglia (DAM) phenotypes, marked by reduced expression of key genes such as Apolipoprotein E, Triggering Receptor Expressed on Myeloid cells 2, Integrin Subunit Alpha X, MER proto-oncogene, tyrosine kinase, Axl, and secreted phosphoprotein 1, particularly in females. It also increases the number of microglia in the hippocampus (in both sexes). This immune impairment is coupled with a decline in NSC maintenance expressing GFAP+ and sex-determining region Y-box 2 in a sex- and region-specific manner. In contrast, neurogenesis is differentially regulated; loss of Atg5 leads to increased Doublecortin neuroblasts and proliferating progenitors in the subventricular zone, indicating an imbalance between stem cell maintenance and differentiation. Orthogonally, Atg5 loss impairs glycolytic capacity (via reduced GLUT1), limiting microglial activation and their ability to support the stem cell niche. 66
The conditional knockout of Atg5 in α-SMA-expressing cancer-associated fibroblasts (CAFs) disrupts autophagy, impairing their ability to process tumor antigens and form immunological synapses with Foxp3+ regulatory T cells (Tregs), thereby reducing Treg activation, proliferation, and tumor infiltration. This leads to inflammatory reprogramming of CAFs, marked by upregulated cytokines/chemokines (e.g., Cxcl5, Cxcl13, Il6, Il18) and enhanced antigen presentation/phagocytosis pathways. Transcriptomic/proteomic analyses reveal downregulated oxidative phosphorylation/respiratory chain genes, including, Cytochrome c oxidase subunit 8A, Ndufa1, and mTOR (mammalian target of rapamycin) signaling (Akt1, Lamtor1), shifting CAFs toward pro-inflammatory, immune-permissive metabolism favoring glycolysis. 88
In another study, ATG5 is part of a ferroptosis-related risk signature where higher expression contributes to poor prognosis in oral squamous cell carcinoma. In high-risk groups, metabolic pathways (e.g., p53 signaling and pyruvate metabolism) are enriched, and immune infiltration and function (e.g., CD8+ T cells, interferon responses) are suppressed, suggesting an immunosuppressed microenvironment. Moreover, both stemness indices (mRNAsi/mDNAsi) are higher in tumors and show positive correlation with the risk score, indicating increased stem-like properties in high-risk conditions. 89
Anti-viral immunity and metabolism in cells are crucially balanced via ATG5-dependent autophagy. SARS-CoV-2 infection results in failure of autophagy, causing amino acids and nucleotides to pile up and feed viral replication, which is worsened by ATG5 knockdown by trapping the nutrient overload. 90 Interferon signaling upregulates diamine acetyltransferase 1, depleting autophagy-essential polyamines (spermidine/spermine) via conversion to putrescine, which impairs eukaryotic translation initiation factor 5A hypusination and transcription factor EB activity. ATG5 loss synergizes with this innate immune paradox, suppressing autophagy further to weaken defenses and enable viral persistence. 91
NLRP3 inflammasome activation is promoted by blocking ATG5-dependent autophagy, the primary mechanism for NLRP3 lysosomal degradation. 92 Without ATG5, autophagosome formation is impaired, and clearance of NLRP3 protein accumulating at the LRR domain is blocked, leading to elevated cleaved Caspase1, IL-1β, and IL-18 secretion in lipopolysaccharide/nigericin-stimulated macrophages. This autophagy collapse disrupts metabolic homeostasis, as evidenced by reduced LC3-II (Microtubule-associated protein -light chain 3-phosphatidylethanolamine conjugate) flux, mitophagy, and lipophagy, causing ROS buildup and lipid dysregulation, thereby amplifying the inflammasome signaling. The resulting pro-inflammatory environment (TNF-α/IL-6/IL-1β) creates a feedforward loop where metabolic stress from impaired autophagy sustains NLRP3 stabilization and immune hyperactivation. 93
Discussion
Although ATG5 is widely recognized as a key part of the autophagy machinery, the evidence reviewed here shows that it has a much broader role. The consistent requirement for ATG5 during organogenesis of the lung, liver, heart, and brain relative to other ATG genes, which show a tissue-restricted pattern, suggests that its functional repertoire is particularly broad. The findings discussed here suggest that this distinctive pattern reflects its roles in stem cell fate determination, cellular metabolic regulation, and immune homeostasis, extending well beyond autophagosome biogenesis. This is highly relevant in the context of embryonic organogenesis, where the developing lung, liver, heart, and brain must undergo changes in cell identity, metabolic potential, and immune competence at multiple time points. All these interdependent functions depend on the coordinated engagement of ATG5 at multiple nodes and cannot be considered modular.
Interestingly, in some stem cell systems, ATG5 promotes differentiation, while in others it supports stemness. We can reconcile these observations by proposing a model in which ATG5 is not a pro-differentiation or a pro-stemness factor, but rather a cellular rheostat that allows execution of whichever program the cell is primed to carry out, determined by the surrounding signals and transcriptional state of the cell at a particular developmental stage of the organism.28,29,32,36 However, a critical unanswered question is what molecular features of the cellular environment drive ATG5 from a stemness-maintaining to a differentiation-promoting factor? Answering this question would make ATG5 easier to study as a practical therapeutic target.
Although a strict causal direction cannot be explicitly defined from current evidence, most evidence thus far favors metabolic regulation as the proximal output of ATG5 activity, followed by downstream events regulating fate and immune programming. This metabolic regulation is accomplished by lipophagy and mitophagy, modulating the bioavailability of metabolic building blocks, acetyl-CoA, for example, that directly modulate gene expression and cell identity via histone acetylation.62,63 This was most clearly observed in CD4+ T cells, where loss of ATG5 resulted in a metabolic switch to glycolysis that directly drives pathogenic Th17 polarization, showing that a metabolic switch alone is capable of driving immune cell fate. 11 Taken together, these observations suggest that there is a self-amplifying loop where metabolic disruption activates inflammation, which further damages mitochondrial and lipid metabolism, worsening all three programs concurrently, as evidenced by the simultaneous disruption of metabolism and inflammation observed in hepatic CD11c+ myeloid cells upon ATG5 loss 71 and further supported by the concurrent dysregulation of stemness, metabolic, and immune programs in our proteomic analysis of Atg5-deficient cells. 80
Most of the work described here has been done using loss-of-function approaches that cannot discriminate between the loss of canonical autophagy and the loss of ATG5′s autophagy-independent functions, which could be mechanistically and therapeutically consequential, considering that targeting one, or both, of these pathways may require fundamentally different approaches. Moreover, another area that needs our attention is sex-specific regulation. As discussed previously, loss of ATG5 disrupts DAM phenotypes and neural stem cell maintenance differently between males and females. 66 This aspect raises a question about the role of sex hormones or sex-specific epigenetic pattern on regulation of ATG5. Hence, future studies must consider gender as a variable while developing ATG5-targeted therapies.
It is the critical importance of ATG5 in human development that is best exemplified by the dose-dependent phenotypes arising from its depletion in humans (Table 2). Ablation of Atg5 in mice results in lethality in early postnatal development, with Atg5-knockout pups dying just 1 day after birth. 96 In addition to this, clinical evidence indicates that this is similarly true in human development. Kim et al. (2016) were among the first to report the deleterious effects of a pathogenic mutation in an essential ATG gene in humans, having identified a homozygous missense mutation (E122D) in ATG5 in two brothers with developmental ataxia, mental retardation, and developmental delay. By interfering with ATG12-ATG5 conjugation and autophagic flux, the partial depletion of ATG5 expression was enough to lead to severe neurodevelopmental defects, with the authors further predicting that full depletion of ATG5 would prove incompatible with human life. 94 These predictions were validated by a later case report involving an infant with compound heterozygosity causing depletion of both ATG5 protein and LC3-II, exhibiting prenatal growth restriction, hydrops fetalis, transfusion-dependent congenital dyserythropoietic anemia, lissencephaly, hypoplasia of the corpus callosum, and death at 5 months of age. Mechanistic investigations into the causes revealed a defect in ATG5-dependent mitophagy during the maturation of red blood cells, as the patient had accumulated undegraded mitochondria within erythrocytes, providing direct proof of the need for mitophagic flux during mammalian hematopoiesis. 95 Taken together, the above examples suggest that the physiological roles of ATG5 outlined in this paper are not exclusive to model systems but have human health implications in accordance with the extent of ATG5 deficiency.
Clinical Evidence Supporting the Developmental Requirement of ATG5 in Humans

From a therapeutic perspective, ATG5 is appealing but complicated. Since ATG5 plays a role in maintaining healthy stem cells, immune tolerance, and metabolic stability, inhibition in the whole body would induce undesirable side effects. In contrast, in cancer stem cells and resistant tumors, the survival benefit associated with ATG5-dependent autophagy can be used for therapeutic gain.44,45 A more practical approach would be to selectively inhibit ATG5-dependent signaling in a tissue-specific manner based on the nature of the disease, stage, and the dynamics of autophagy flux and canonical versus noncanonical activity of ATG5.
In summary, the data presented here point to an emerging paradigm where ATG5 serves as a context-specific regulator of cellular remodeling processes that coordinate stem cell development, metabolic fitness, and immune responses during development and disease.
Footnotes
Acknowledgments
The authors acknowledge the facility support from DST-FIST, K-FIST, TIFAC-CORE, DBT-BUILDER, VGST Karnataka, Manipal School of Life Sciences, and Manipal Academy of Higher Education, Manipal, Karnataka, India. S.M. acknowledges the NYU Langone Grossman School of Medicine, Department of Radiation Oncology, New York, NY 10016, United States, where he is involved as a sponsored individual without any financial interest. They apologize to the authors whose findings could not be cited in this work due to space limitation.
Author Disclosure Statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Funding Information
Work in the S.M. lab is supported by funding from the Anusandhan National Research Foundation—Advanced Research Grant (
Supplemental Material
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.