
Abstract
Aging is a complex process characterized by the accumulation of molecular damage that leads to cellular dysfunction and tissue deterioration. Among the various types of contributing molecular damage, aberrant protein crosslinks are recognized as a key contributor to age-related pathologies. Crosslinks occurring at lysine and arginine residues, such as advanced glycation end-products (AGEs) and carbamylation, have attracted considerable attention of the aging research community. In contrast, the roles of cysteine-derived crosslinks in aging pathobiology remain underappreciated. While native disulfide formation is essential for protein structure and function, the same redox features that make cysteine indispensable to protein biochemistry also render it particularly susceptible to nonspecific disulfide crosslinking. The body exploits specialized protective thiols such as glutathione to maintain redox homeostasis and counteract the deleterious effects of aberrant disulfide formation. However, these endogenous protective measures decline with aging, resulting in the accumulation of oxidative cysteine modifications. In this review, we highlight the emergent roles of cysteine-related molecular damage in age-related disease. Drawing inspiration from endogenous protective thiols, we survey progress in the development of small-molecule therapeutic thiols that show promise in mitigating damage caused by the accumulation of cysteine-derived crosslinks. Understanding the relationship between these aberrant crosslinks and protective thiol interventions in aging diseases, as well as how therapeutic thiols can be improved, is critical for the development of comprehensive treatments.
Keywords
Introduction
Cysteine (CSH) is the least abundant amino acid in proteins, accounting for less than 3% of the total amino acid composition in the human proteome. 1 Yet, despite this scarcity, CSH is essential to both protein function and the endogenous mechanisms that maintain redox homeostasis. Owing to its reactive thiol group, CSH residues form disulfide bonds that stabilize protein structure and allow for regulation of protein activity.2–4 Consequently, cells dedicate significant resources to prevent nonspecific thiol oxidation reactions. Structural disulfide bonds typically exist as intramolecular linkages, and their formation is tightly regulated by specialized enzymes like oxidoreductases, thiol oxidases, and protein folding chaperones. 3 While disulfides generally protect against CSH over-oxidation, their aberrant accumulation is linked to pathogenic aggregation events. 5 In addition, CSH residues undergo other oxidative posttranslational modifications (i.e., ox-PTMs) that can contribute to age-related molecular damage. These effects can be direct, or in certain cases, they can promote the spontaneous formation of aberrant disulfide linkages.
Humans have evolved protective mechanisms to correct aberrant disulfides and maintain redox homeostasis. 6 These corrective measures rely on the low-molecular weight (LMW) thiol glutathione (GSH) and its integration with redox enzymes including NADPH-dependent GSH reductase. 7 Aging is associated with increased oxidative stress and a gradual shift toward an oxidative cellular environment.8–10 These changes degrade endogenous antioxidant defenses, thereby promoting the increased formation of stochastic disulfides and other forms of oxidative damage at CSH residues. 11 Several age-related neurodegenerative and cardiovascular diseases are associated with a decline in protective LMW thiols. 12 For example, Parkinson’s disease (PD) is driven by a decrease in GSH levels in damaged regions of the brain. 12 The growing importance of endogenous protective thiols in aging has sparked renewed interest in the design and/or repurposing of thiol-containing therapeutics, many of which have been applied in a limited fashion for decades.13–16 Both FDA-approved thiol-based drugs and biogenic protective thiols continue to be explored as treatments in other areas of disease, with the goal of realizing their potential. Inspired by this emergent research area, this review focuses on the causative role(s) of aberrant disulfide crosslinks in aging pathobiology. With an eye toward intervention, the potential of therapeutic thiols in mitigating the damage associated with age-related diseases is also examined.
Fundamentals of CSH Chemistry
The thiol functional group in CSH is responsible for the specialized features of this unique amino acid. Despite being grouped with oxygen in the periodic table, sulfur is less electronegative and able to accommodate a wide range of oxidation states. 17 As a functional consequence, the thiol S–H bond in CSH (BDE = 82 kcal/mol) is weaker and thus more prone homolytic cleavage than O–H, N–H, and C–H bonds present in proteins. The S–H bond is also more acidic than many other functional groups in biochemistry. The pKa of free CSH is 8.5, which allows for a low concentration of the corresponding nucleophilic thiolate anion at physiological pH. When embedded in proteins, the pKa of CSH residues can be lowered through interactions with nearby residues positioned to stabilize the thiolate anion. Cumulatively, these properties enable a range of one-electron and two-electron reactions involving the thiol group. Thus, CSH is exploited as a reactive residue in enzyme active sites, as a preferred site for posttranslational modifications (e.g., prenylation), and as a ligand for metals (e.g., Zn and Fe). The biochemistry of CSH has been reviewed.7,18 As such, this review focuses on reversible and irreversible oxidation reactions of CSH and their contributions to age-related disease.
Thiol/disulfide fundamentals
As outlined above, cells expend considerable energy to prevent nonspecific oxidation at CSH residues. Thus, the formation of structural disulfide bonds in proteins is mediated by specialized enzymes within dedicated cellular compartments including endoplasmic reticulum, mitochondrial membrane space, and periplasmic space. In healthy cells, the cytosol and nucleus maintain a reducing environment to prevent thiol oxidation. However, as oxidative stress increases with aging, the accumulation of reactive oxygen species (ROS) in the cytosol can promote nonspecific disulfide formation via several different pathways. As summarized in Scheme 1, CSH residues undergo auto-oxidation to form disulfides (i.e., cystine or CSSC). One-electron oxidation of the thiol group, or the corresponding thiolate, generates a thiyl radical (CS•). This sulfur-centered radical gives rise to CSH modifications including S-nitrosothiols and persulfides that react spontaneously and nonspecifically with nearby CSH residues to form stochastic disulfides. Similarly, two-electron oxidation of CSH thiols by hydrogen peroxide (H2O2)—a common ROS—affords a sulfenic acid intermediate. This transient species captures nearby thiols to generate disulfides with concomitant loss of water. Nonspecific disulfide formation at structural or functional CSH residues can impact enzymatic activity, protein folding, protein-protein interactions, and protein turnover. 19 Similarly, disulfides formed between proteins can promote aggregation events that contribute to disease pathobiology.
Owing to the weak S–S bond, disulfides can be reduced to regenerate free thiols. Thus, in theory, molecular damage caused by aberrant disulfides is reversible. This process is not always direct. For example, disulfides react with thiolate anions to form mixed disulfides, which are reduced further by redox enzymes to regenerate free thiols. Such thiol/disulfide exchange reactions are involved in the activity of protein disulfide isomerases (PDIs), a class of enzymes that correct aberrant disulfides in misfolded proteins. 20 This process may also be involved in the S-glutathionylation of CSH residues, wherein a mixed disulfide is formed between CSH and GSH. The reversible formation of GSH-mixed disulfides protects reactive CSH residues from oxidative damage. Intriguingly, only 0.1% of total CSH residues are S-glutathionylated in nonstressed cells. 21 This population can increase up to 15% under oxidative stress conditions, thus highlighting a distinction between disulfides that contribute to protein dysfunction and mixed disulfides formed with GSH that serve a protective role.
Oxidative PTMs
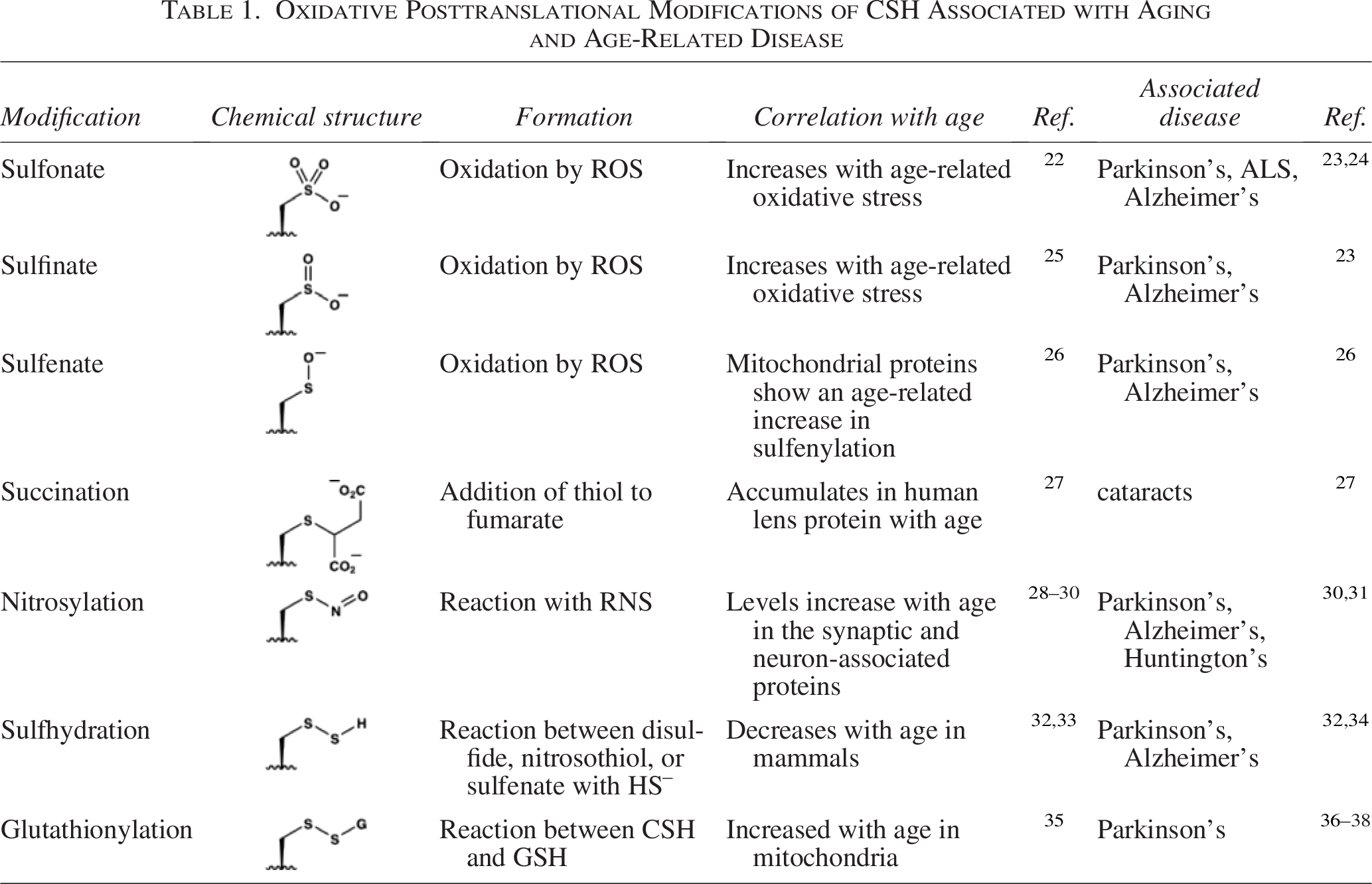
Disulfides represent one of many oxidative modifications of CSH residues. This topic has received considerable attention, particularly in the context of oxidative posttranslational modifications (ox-PTMs) and their roles in the regulation of protein function. 17 Most ox-PTMs at CSH residues are reversible, allowing them to function as redox switches to regulate cell signaling and localization. However, oxidative stress can lead to accumulation of reversible and irreversible ox-PTM products. Those directly associated with age-related diseases are outlined in Table 1. These ox-PTMs are often triggered by the direct reaction of the thiol groups with ROS, reactive nitrogen species (RNS), electrophilic metabolites, and as mentioned above, other thiols to generate mixed disulfides (e.g., S-glutathionylation). 39
Oxidative Posttranslational Modifications of CSH Associated with Aging and Age-Related Disease
Endogenous Protective Thiols
Protective mechanisms manage the constant generation of cellular ROS and RNS, which if left unregulated, can promote oxidative damage to DNA, lipids, and proteins. 40 LMW thiols are integral to these defenses. Organisms maintain millimolar concentrations of these protective small molecules that function as cofactors for redox enzymes, form mixed disulfides with CSH residues in proteins to protect against overoxidation, scavenge free radicals, and reduce ox-PTMs in proteins. Despite its redox activity, free CSH is not exploited as a protective thiol on its own. Instead, CSH is integrated into more complex structures with refined properties and scaffolding that serve as recognition elements for enzymes and other biomolecules within the cell.
Glutathione
The tripeptide γ-glutamylcysteinyl glycine (GSH) serves as the main regulator of redox homeostasis in mammalian cells. 41 The CSH residue embedded within GSH is responsible for its redox activity; however, relative to CSH, the functional thiol group in GSH is both less acidic (pKa = 8.9) and a more powerful reductant (reduction potential, E° = –240 mV). Consequently, only 2.9% of GSH is present as the reactive thiolate in the cytosol. Moreover, the structure of GSH contains an atypical γ-glutamyl peptide linkage that allows specific integration with GSH-dependent enzymes. As shown in Figure 1A, GSH reacts with ROS like H2O2 and free radicals to generate the corresponding disulfide (i.e., GSSG). While physiologically relevant, the rates of such reactions are 3–5 orders of magnitude slower than those of dedicated antioxidant enzymes. 42 Thus, GSH serves as a reducing equivalent, particularly for thiol peroxidases that reduce intracellular ROS such as GSH peroxidases and peroxiredoxins. 43
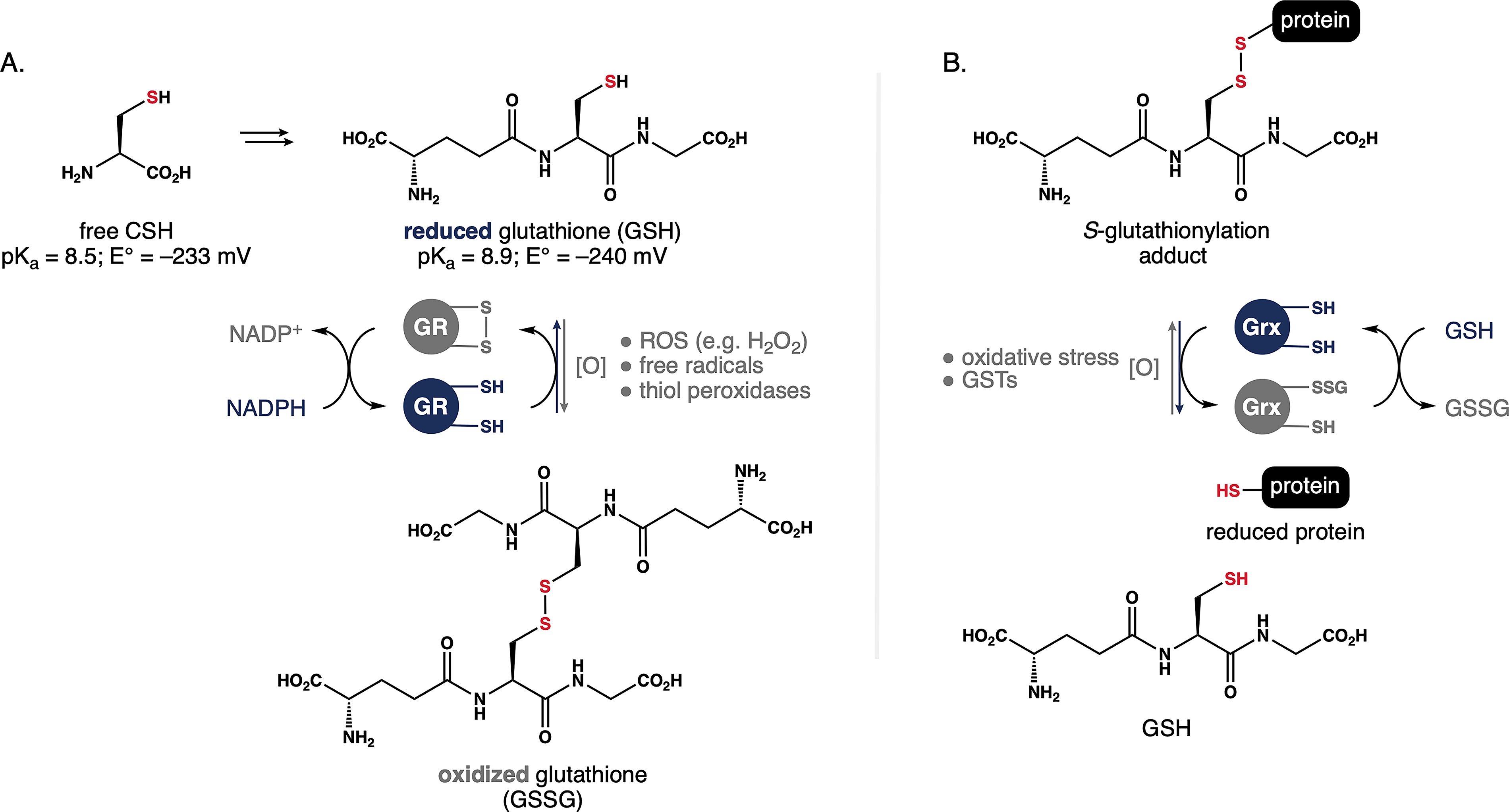
Mechanisms regulating the GSH/GSSG redox buffer.
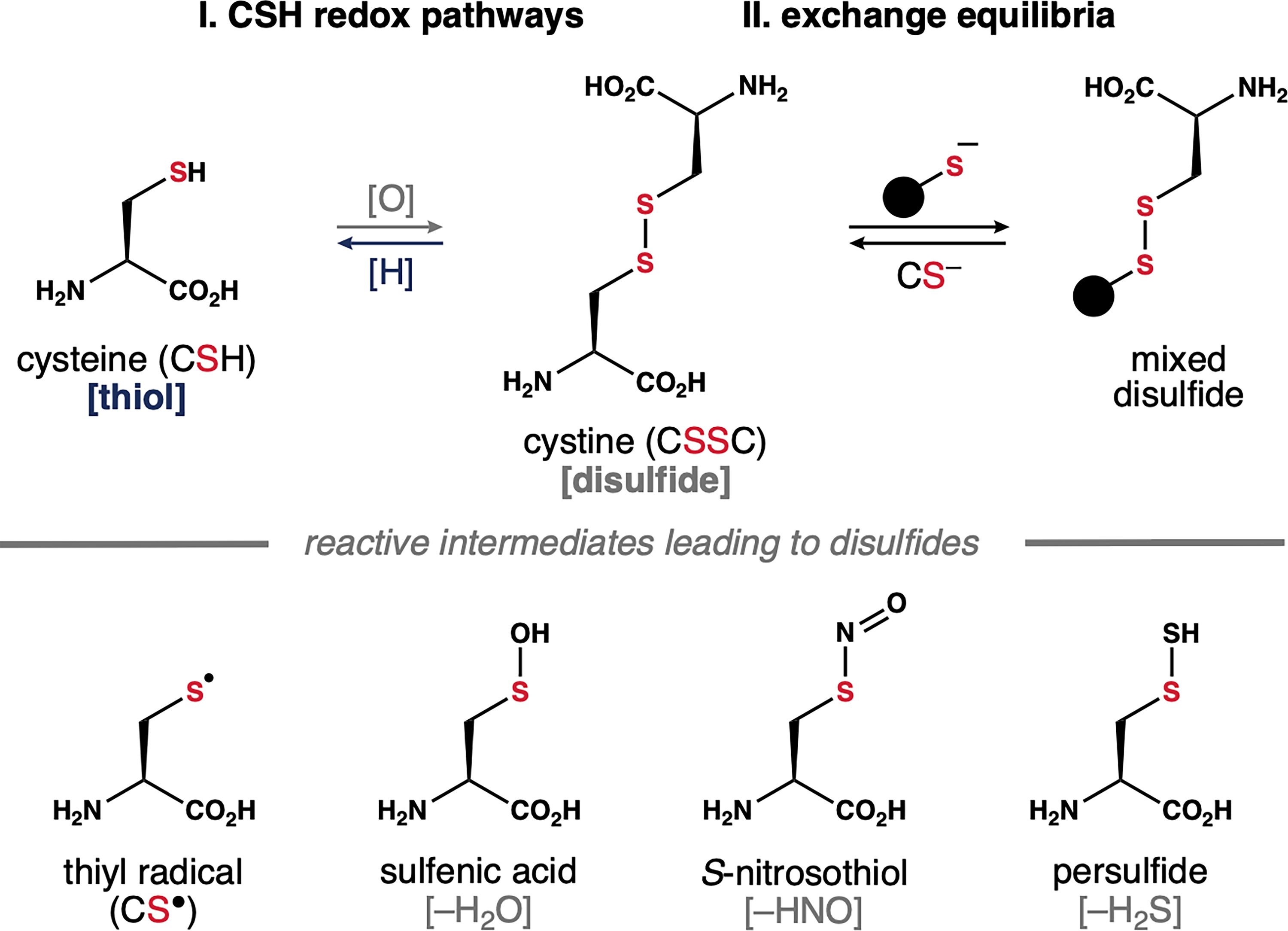
Fundamentals of thiol/disulfide dynamics.
Concentrations of GSH and GSSG are tightly regulated within different cellular compartments to generate a localized “redox buffer.” The cytosolic GSH:GSSG ratio in normal cells is typically >100:1 and maintained by glutathione reductase (GR), a FAD-dependent enzyme that transfers electrons from NADPH to GSSG to (re)generate two molecules GSH. This NADPH-dependent redox couple is central to many of the endogenous mechanisms that repair disulfide bonds. These include PDI, a chaperone enzyme that regulates protein folding in the endoplasmic reticulum by catalyzing the formation of native disulfide pairing. 20 More recently, PDI has gained attention from the aging community for its ability to repair double-stranded DNA lesions. 44 In both cases, the GSH/GR redox couple plays an essential role in reducing the catalytic CSH/CSSC center in the PDI active site.
In an oxidative environment, the GSH:GSSG ratio rapidly decreases and GSH is consumed faster than it is produced. The thiol in GSH can form the same reactive intermediates described for CSH in Scheme 1. These can induce spontaneous S-glutathionylation of available CSH residues to regulate protein function in response to oxidative stress (Fig. 1B). Alternatively, GSH is added to reactive CSH residues by glutathione S-transferases (GSTs). 45 This posttranslational modification protects against thiol reactivity and protein misfolding. In either event, the resulting mixed disulfide can be cleaved by glutaredoxin (Grx) in a process that liberates GSSG, ultimately allowing the liberation of GSH through the action of GR. In this way, cells use S-glutathionylation as both a protective measure against oxidative stress and means to store GSH reserves.
Alternative protective thiols
While GSH functions as the prototype protective thiol in human biology, many organisms have evolved alternative LMW thiol cofactors to regulate cellular redox homeostasis (Fig. 2). For example, certain bacteria utilize coenzyme A (CoA) as an alternative reducing equivalent. 46 While CoA (E° = 234 mV) has a similar reduction potential to GSH, its S–H bond is considerably less acidic (pKa = 9.8), and thus, it is present exclusively as thiol at physiological pH. In analogy to GSH, the oxidized CoA disulfide is reduced in an NAD(P)H-dependent fashion by CoA disulfide reductase (CoADR). 47 The glucosamine/CSH adducts mycothiol (MSH) and bacillithiol (BSH) function as GSH surrogates in actinomycetes and Gram-positive bacteria, respectively.48,49 Although the roles of these metabolites extend beyond redox homeostasis, they have function as redox couples similar to GSH/GR. A variety of marine invertebrates leverage ovothiol A (OSH) to protect against ROS. 50 This chemotype is especially effective at scavenging ROS due to the low pKa of the thiohistidine group. Under physiological conditions, OSH is exclusively present as the thiolate. By comparison, the isomeric thiohistidine of ergothioneine (ESH) is less reactive because it exists between its tautomeric thiol and thione forms. 51 As a result, ESH is often produced in combination with other LMW thiols, which are required as complementary reductants. 52 Nevertheless, the sulfur atom in ESH is redox-active and serves as an effective antioxidant against ROS, such as singlet oxygen and hydroxyl radicals.
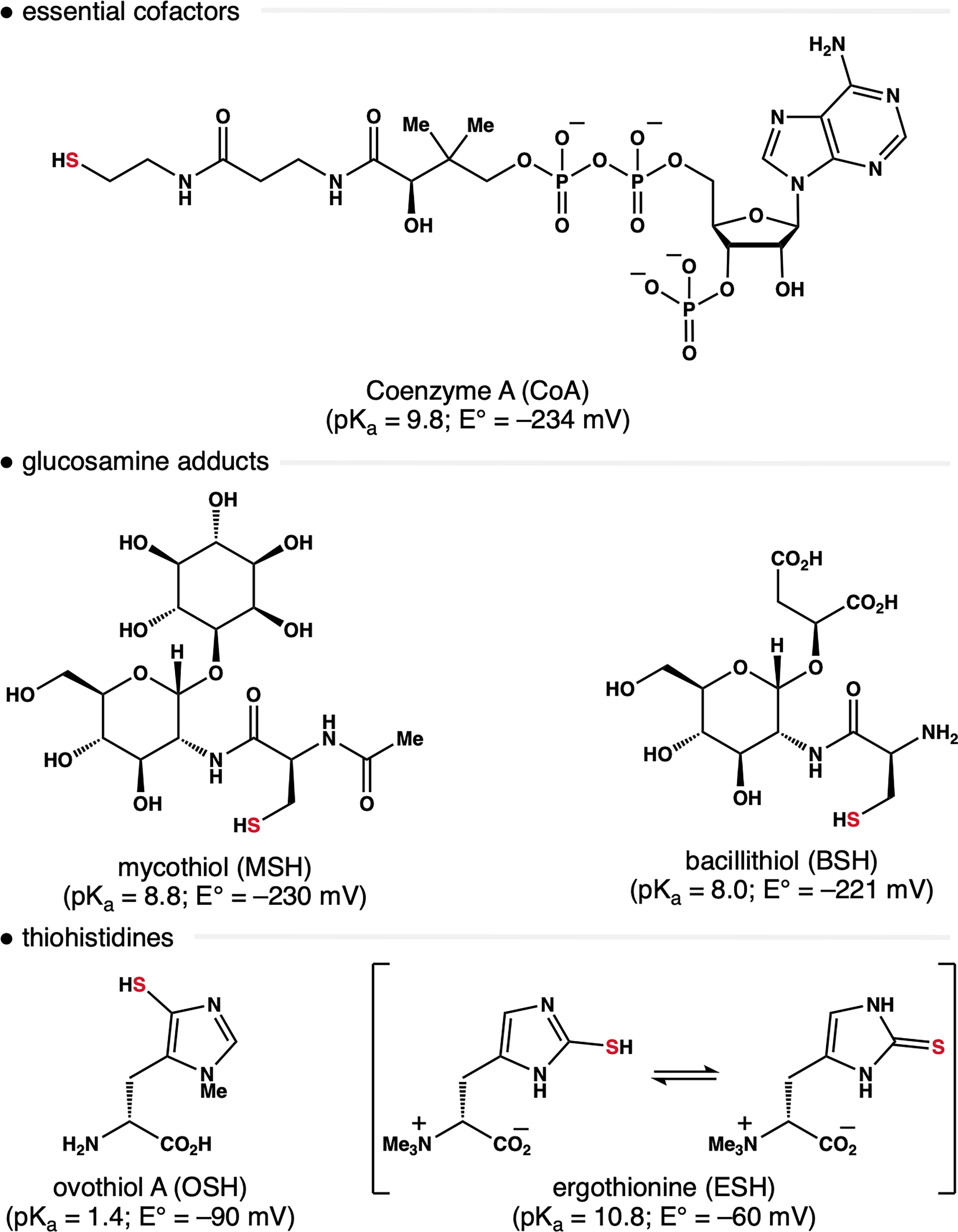
Alternative protective thiols that contribute to redox homeostasis in nonmammalian cells. In each case, the redox-active thiol is ultimately derived from metabolism of CSH.
Despite their structural differences, in analogy to GSH, the redox-active sulfur atom in other LMW thiol antioxidants is derived from CSH. This relationship is nonobvious in the thiohistidines OSH and ESH. In both cases, the thiol group is transferred from CSH to the imidazole ring by enzymes. 51 Thus, it appears that diverse organisms have evolved disparate biosynthetic pathways to convert CSH into a reducing equivalent that is tailored to the needs of their environment. The preferred status of CSH as a progenitor to endogenous protective thiols, as well as the different evolutionary strategies to refine the reactivity of the S–H bond, may inform the design of thiol-based therapeutics.
Aberrant Disulfides and Oxidative Stress
Native disulfide bonds in proteins are often classified as structural or redox-sensitive. Structural disulfides are required for proper folding and function of many proteins. They exist as either intra- or interdomain linkages and are often installed by folding chaperones (e.g., PDI) as part of the protein folding process. 3 In contrast, redox-sensitive disulfides are generally more reactive and localized within the exposed regions of proteins.
Stochastic disulfides form in both structural and redox-active systems as oxidative stress in the cell increases. In static systems, stochastic disulfides can be attributed to the lack of a specific folding mechanism. Thus, they are common in misfolded or partially unfolded protein states but are often corrected by repair mechanisms to the native state. 53 In general, surface-exposed CSH residues are the most accessible to oxidizing agents and thus prone to form stochastic disulfides. 19 These bonds occur frequently at redox-sensitive CSH residues due to fluctuating redox conditions, and at low levels such linkages function as homeostatic switches. 54 However, when oxidative stress becomes more extreme, reversible stochastic disulfide formation can introduce aberrant disulfide crosslinks with implications arising from protein misfolding, aggregation, or cellular dysfunction.55–57 Aberrant disulfides represent a subclass of stochastic linkages where incorrect CSH pairing leads to a major disruption of protein function. These crosslinks can also result from mutations that introduce or remove CSH residues. In either event, stochastic disulfide formation is associated with disrupted cell growth and, eventually, induction of apoptosis. 54
Aging is associated with a decline in mitochondria efficiency. 58 This results in less ATP production, elevated ROS, and a decline in the GSH:GSSG buffer ratio. The slow accumulation of oxidative stress triggers damage to cells resulting in dysregulation of protein function, changes in cell signaling, and the onset of cell senescence. 54 Under these conditions, CSH residues are prone to aberrant disulfide bond formation and damage caused by other oxPTMs that disrupt protein structure and function. At the same time, oxidative changes to the cellular microenvironment and redox potential landscape disrupt corrective refolding of proteins, which as outlined above, is dependent on GSH:GSSG redox buffer. Misfolded proteins can bring CSH residues that were originally far apart into proximity, thereby inducing aberrant disulfides and protein aggregation.59,60
It is now established that oxidative protein damage accumulates exponentially with age. 61 Thus, aging and age-related diseases can be viewed as emerging phenotypes of progressive proteome damage, including malfunction as a result of accumulated aberrant CSH crosslinks. 62 In addition to oxPTMs of CSH, irreversible covalent modifications such as AGEs and advanced lipoxidation end products (ALEs) also drive age-related diseases. 63 In analogy to AGEs, aberrant disulfides drive pathology in both direct and indirect ways. Both damage proteins at a structural level. Similarly, while AGEs indirectly cause inflammation via AGE receptors, aberrant CSH modifications disrupt cellular signaling by occupying redox-active CSH residues. However, unlike AGEs and ALEs that are irreversible chemical linkages, most oxidative CSH modifications are reversible. This fundamental difference provides an opportunity to minimize—and even fully correct—the detrimental effects of aberrant disulfides through the rational design of therapeutics.
CSH-Based Oxidative Damage in the Pathobiology of Age-Related Diseases
The interplay between aging, oxidative stress, and reversible oxidative damage at CSH residues can have a significant impact on protein function. As such, direct connections between CSH-based oxidative damage to proteins and the pathobiology of age-related diseases continue to emerge. The formation of β-sheet-rich fibrils (i.e., amyloids) from native protein structure is perhaps the most recognizable example. Disulfide bonds are critical to amyloid formation, 64 a process central to many age-related disorders including Alzheimer’s disease, PD, amyotrophic lateral sclerosis (ALS), and Type II diabetes. Whereas intramolecular disulfides impart stability to native protein structure and decrease the tendency for amyloid aggregation, stochastic disulfides promote aggregation events that drive amyloid diseases. 18 Similarly, a novel form of cell death termed disulfidptosis has gained attention in the field of ischemic stroke research.65,66 This process, which is distinct from programmed cell death, involves abnormal CSH accumulation and aberrant disulfide bond formation in actin cytoskeleton proteins. 65
An overview of the literature reveals that oxidative modifications at CSH residues contribute to damage that falls within the glycoSENS, amyloSENS, and lysoSENS categories of aging damage. 67 Examples highlighting a connection between well-characterized CSH-based oxidative modifications and tangible protein damage driving aging pathologies are summarized below:
Marfan syndrome
Fibulin-3 (FBLN3) is a CSH-rich glycoprotein involved in the organization of the extracellular matrix (ECM). FBLN3 contains six growth factor domains, each containing three conserved disulfide bonds. Whole exome sequencing of siblings with Marfan syndrome 68 —a genetic disorder that affects connective tissue—revealed the homozygous CSH missense mutation C55R, which acts indirectly by leaving its disulfide partner C70 available on adjacent FBLN3 monomers. This genetic change induces the formation of extracellular disulfide-linked FBLN3 dimers that promote aggregation and render the protein inactive. 69
Alzheimer’s disease
This neurodegenerative disorder characterized by the accumulation of amyloid-β plaques and neurofibrillary tangles composed of hyperphosphorylated tau protein. The amyloid-β peptide (Aβ) is generated when the amyloid precursor protein is cleaved by β-site amyloid precursor protein cleaving enzyme 1 and γ-secretase. The release of Aβ is associated with increased oxidative stress, which promotes aberrant disulfide-linked aggregates of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). 70 These Aβ-induced aggregates of GAPDH form toxic mixed aggregates with Aβ itself that accelerate amyloidogenesis and neuronal death. Neuroinflammation caused by the accumulation of Aβ aggregates also triggers excessive nitric oxide (NO) production via induction of neuronal NO synthase. This RNS species promotes aberrant S-nitrosylation leading to defects in glutaminergic signaling and thus hyperexcitability and synaptic damage. 71
Parkinson’s disease
PD is a neurodegenerative disease characterized by the formation of Lewy bodies and the death of dopaminergic neurons. 72 Oxidative stress is involved in the pathology of PD, and mutations within the antioxidant protein DJ-1 correlate with early onset of PD. 73 Sulfenic acid formation or S-glutathionylation of C106 in DJ-1, as well as mutations to this residue, increase sensitivity to the neurotoxin 1-methyl-4-phenylpyridinium. Genetic knockdown of Grx with si-RNA decreases levels of DJ-1, suggesting that deglutathionylation of DJ-1 protects this protein from degradation. 36 In addition, Grx levels are reduced in dopaminergic neurons of PD patients. 37 The overexpression of DJ-1 in a C. elegans model of PD containing a knockdown homolog of Grx (i.e., GLRX-10) led to dopaminergic neuronal protection. Taken together, these data indicate that aberrant S-glutathionylation of DJ-1 contributes to PD. 38
Amyotrophic lateral sclerosis
Certain forms of this inherited disease are caused by mutations in superoxide dismutase 1 (SOD1) results in protein misfolding and aggregation into insoluble cross-β-amyloid fibrils that cause cell death. In ALS, SOD1’s critical homodimeric disulfide affects aggregation kinetics. Whereas reduced SOD1 shows increased fibrillation rates, oxidized SOD1 resists fibril initiation. 74 Interestingly, a disulfide mutant of SOD1 was more readily reduced than wild-type (wt) SOD1, and small quantities of reduced mutant initiated fibrillation of wt-SOD1. 75 These data implicate the disulfide bond of SOD1 as an important contributor to the aggregation kinetics in ALS.
Arteriosclerosis
Arteriosclerosis is characterized by the hardening of the arteries and is the primary cause of coronary artery disease. Atherosclerosis, a specific class of arteriosclerosis, is characterized by the deposit of plaque composed of lipids. Through comparative genome analysis, apolipoprotein A-V (ApoA-V) was found to modulate plasma triacylglycerol homeostasis. 76 As a regulatory plasma protein, ApoA-V associates with low- and high-density lipoprotein to promote lipoprotein lipase-mediated triacylglycerol hydrolysis. The preprotein of ApoA-V, APOA5, contains single-nucleotide polymorphisms (SNPs) that correlate with increased plasma triacylglycerol. 77 Among those SNPs, G185C of APOA5 corresponds to G162C in mature Apo-V. 78 The introduction of an additional CSH residue in Apo-V leads to disulfide-linked heterodimer formation with the nearby residue C204. As a result, this disrupts ApoA-V–lipoprotein binding and triacylglycerol homeostasis, thereby causing the arterial walls to harden and lose elasticity. 76
Presbyopia and cataracts
Presbyopia is characterized by loss of near vision and focus ability while cataracts is characterized by clouding and opacification of the lens. The eye lens is rich in long-lived crystallin proteins, which are essential for maintaining lens transparency and refractive power. As long-lived proteins, they are susceptible to oxidative damage over time as GSH levels decrease and redox homeostasis is disrupted. While the exact mechanism behind presbyopia and cataracts has yet to be elucidated, age-related accumulation of disulfide-induced insolubility plays a central role in lens transparency and elasticity.79–81 In human γS crystallin, the solvent-exposed cysteine triad of C22, C24, and C26 undergoes a complex set of disulfide exchange reactions under a simulated lens environment found during cataract formation. This highlights the susceptibility of crystallin proteins to oxidation and their importance as a protective oxidative sink. Such modifications reduce protein solubility and may contribute to lens stiffness relevant to presbyopia. They also cause γS crystallin to aggregate if surface-exposed disulfides are transferred to buried cysteines. In human cataractous lenses, at least 80% of the cysteine residues C170 (βA1/3) and C83 (γS) are oxidized, suggesting that these CSH residues play a role in early age-related nuclear cataract. 82
Sorsby fundus dystrophy
This is a rare progressive autosomal dominant disease that is characterized by retinal atrophy and detachment leading to blindness. Disease onset occurs in the third decade of life and loss of central vision from macular atrophy comes in the fifth. 83 The Sorsby fundus dystrophy locus contains the gene timp3 encoding tissue inhibitor of metalloproteinase-3 (TIMP-3). This TIMP-family ECM protein regulates matrix metalloproteinases and plays a pivotal role in determining the extent of matrix degradation during tissue remodeling and ECM homeostasis. 84 Three mutations of TIMP3 in patients lead to a replacement of protein residues by CSH (i.e., S181C, S156C, W172C). When present, these mutations enable the formation of aberrant disulfides that disrupt protein structure and ultimately contribute to macular degeneration. 83
SARS-CoV-2 (COVID-19)
SARS-CoV-2 is a viral infection that causes severe respiratory distress syndrome. 8 The risk of mortality is low for young people, yet it becomes dramatically higher with age. While the exact cause of this disparity remains elusive, a decline in the extracellular CSH:GSSG is one molecular indicator of increased risk of infection and the development of severe symptoms.8,11,85,86 The most severe complications of COVID-19 infection have been associated with a decline in both GSH:GSSG and protein thiol/disulfide ratios in human plasma especially, those patients with pre-existing conditions.9,87,88 A similar decline was observed in the extracellular fluids of the respiratory tract in association with other pulmonary diseases such as idiopathic pulmonary fibrosis (IPF), an age-related disease of the lung caused by oxidative stress, where a four-fold decrease in GSH levels were observed in IPF patients compared with healthy subjects. 89
Thrombotic microangiopathy
Thrombotic microangiopathy (TMA) encompasses a group of disorders characterized by microvascular blood clots inside capillaries and arterioles. Thrombotic thrombocytopenic purpura is a life-threatening disease categorized under TMA that involves platelet adhesion to ultra-large von Willebrand factor (vWF) multimers, affecting the liver, kidneys, heart, pancreas, and brain. 90 The accumulation of these multimers is caused by a deficiency in the vWF-cleaving protease ADAMTS13. 91 Multimer formation is seeded by homodimers formed via disulfide bonding at the C- and N-termini. 92 Biochemical manipulation and spectroscopic analyses of a vWF model construct implicated C1099 and C1142 as key residues in vWF multimer assembly. 93 The key mutations C1099A and C1142A prevented multimer formation, demonstrating that residues C1099 and C1142 are essential to the mechanism of vWF multimerization. 94
Therapeutic Thiols
In recent years, the motivation to repurpose approved drugs 95 has renewed the interest in thiol-based drugs and inspired new development. Therapeutic thiols can enhance endogenous defenses and provide health benefits in diseases that arise from oxidative stress. 96 The effects of therapeutic thiols fall into one or more categories: mucolytics, antioxidants, radioprotectants, anti-inflammatory agents, antibiotics, chelating agents, and disulfide cleaving agents. Phenotypes induced by these structures are often attributed to the redox-active thiol group, which serves as a multifaceted metal chelator (i.e., Hg, Cu, Pb, As), ROS/RNS scavenger, nucleophile, and in some cases, as feedstock boosting GSH biosynthesis. 13 Several reviews on therapeutic thiols, including their limitations, have been published in recent years.13,15,16,97,98 This section focuses on therapeutic thiols that (1) have established utility in combating age-related diseases, and (2) offer insights into the strategic design of next-generation thiol-based drugs. A list of other investigational therapeutic thiols and associated prodrugs that are beyond the scope of this discussion is provided in Table 2.
Select Investigational Thiol Therapeutics and Associated Prodrugs
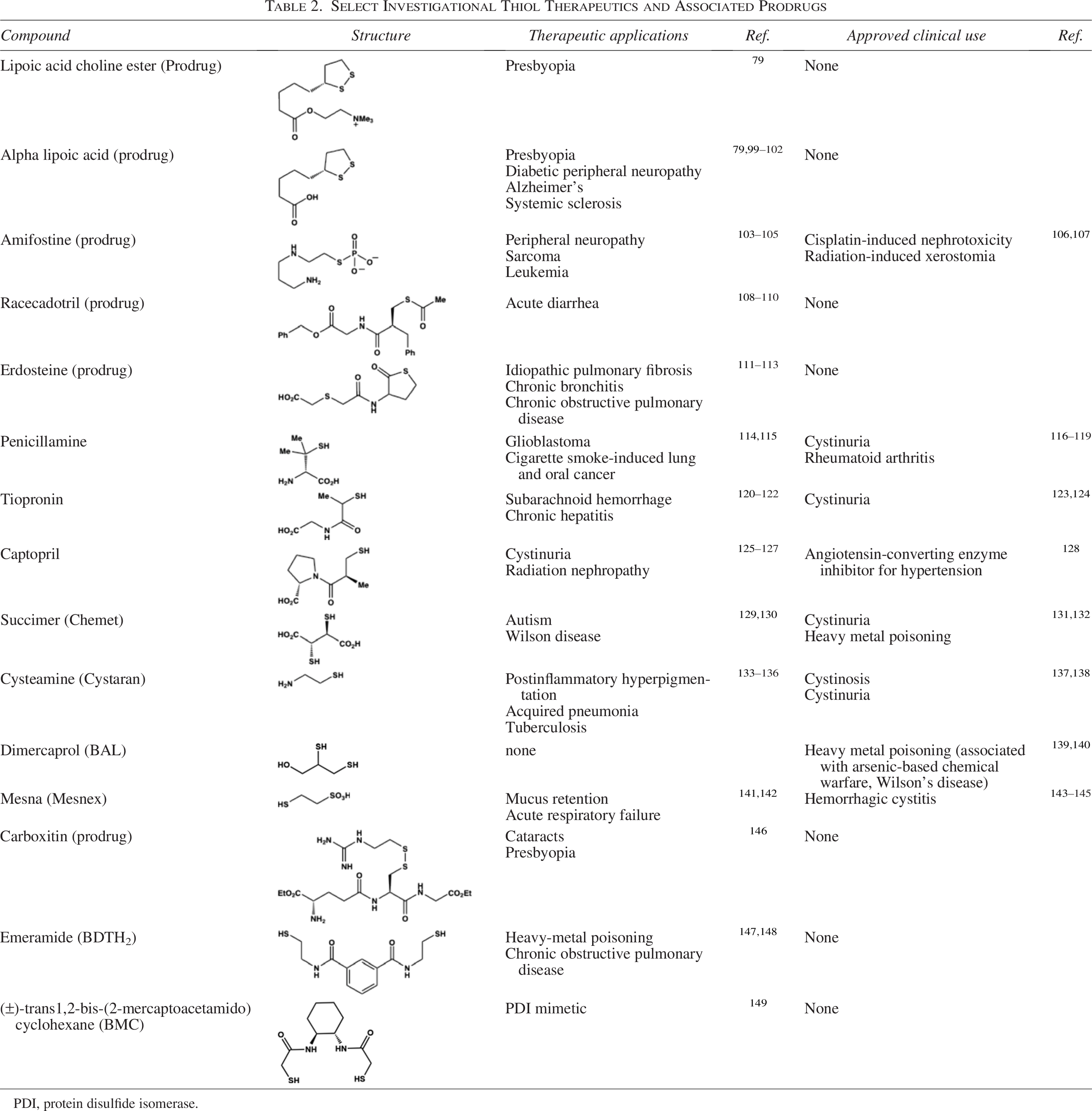
PDI, protein disulfide isomerase.
CSH derivatives
Given the central role of CSH in thiol biochemistry and its integration into protective thiol natural products, it is perhaps not surprising that the CSH scaffold has been explored extensively as a therapeutic. The most notable variant is N-acetylcysteine (NAC, Fig. 3), a CSH derivative that has been used since the 1960s as a mucolytic and acetaminophen overdose therapy. 13 NAC shows promise in several age-related pathologies including reducing inflammation in atherosclerosis,150,151 improving left ventricle function post-MI,152,153 boosting GSH biosynthesis in PD154,155 and AD,156,157 inhibiting diabetic cataract progression, 158 and mitigating oxidative damage in age-related macular degeneration. 159 Several disease models have demonstrated that long-term NAC supplementation can extend lifespan. For example, NAC induced an increase in both median and maximum lifespan in mice, and supplementation of glycine and NAC together (i.e., GlyNAC) increased lifespan even further.160,161
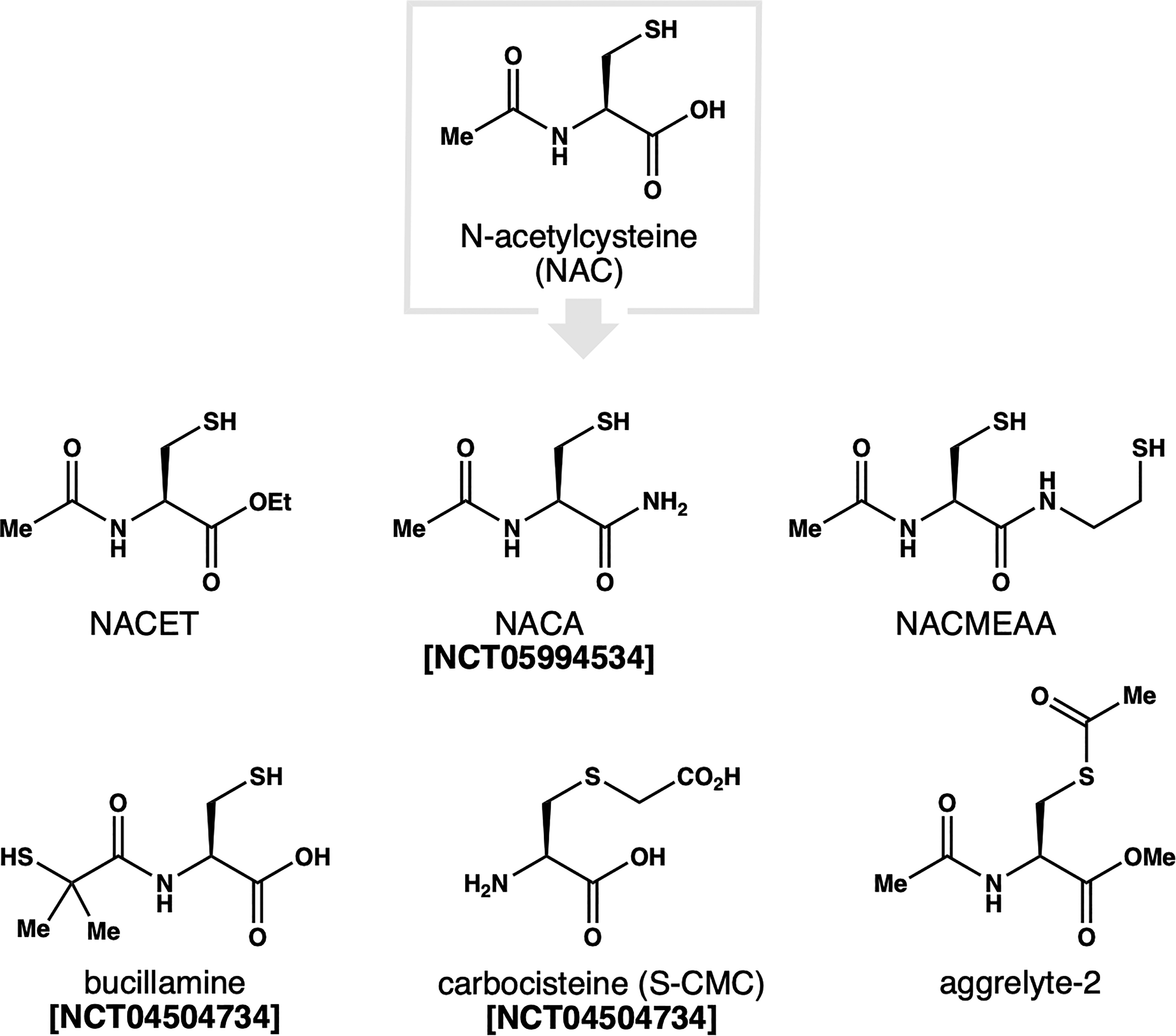
N-acetylcysteine (NAC) and congeners investigated as therapy for age-related diseases.
Although NAC has been popularized as an antioxidant, and applied as such in a range of clinical, animal, and cellular experiments, many questions and misconceptions surround its mechanism of action. For example, NAC is described as a ROS scavenger; however, the rates of reaction between NAC and common oxidants such as H2O2 and superoxide (O2•–) are too slow to be physiologically relevant at any therapeutic concentration of NAC. 162 Instead, NAC primarily acts indirectly by replenishing GSH. This mechanism underlies the clinical use of NAC in acetaminophen poisoning, where it restores GSH levels in the liver that are exhausted by the toxic metabolite N-acetyl-p-benzoquinone imine.163–165 NAC also directly reduces disulfides, as seen in its mucolytic effect or for example its ability to reduce albumin. 152 These activities notwithstanding, the clinical utility of NAC in age-related diseases continues to be debated. 166 As summarized in Table 3, NAC has been investigated in several clinical trials over the past 25 years with mixed results.167–169 Systematic reviews and meta-analyses of these clinical trial data have reached conflicting conclusions on the therapeutic benefits of NAC.170,171
Select Clinical Trials of NAC in Age-Related Diseases from 2000 to 2025
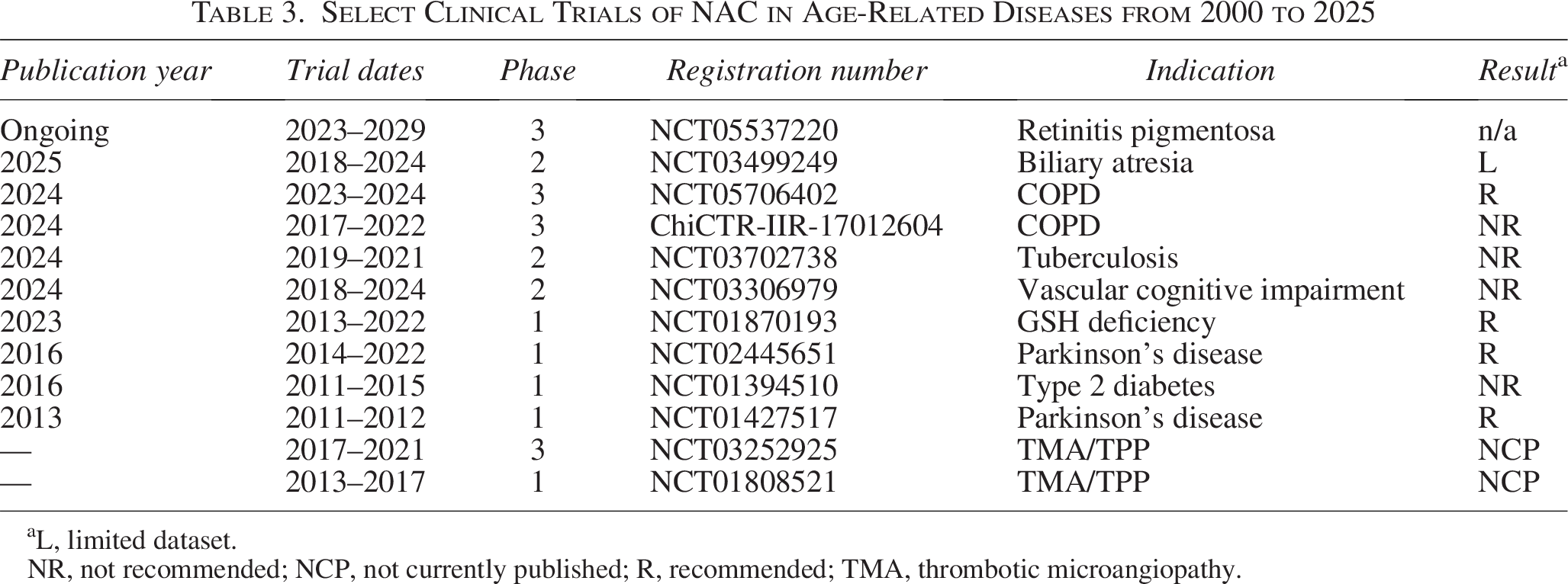
L, limited dataset.
NR, not recommended; NCP, not currently published; R, recommended; TMA, thrombotic microangiopathy.
The physicochemical limitations of NAC have inspired the development of several NAC variants with improved pharmacological profiles. These include NAC ethyl ester (i.e., NACET) and amide (i.e., NACA) prodrugs that enhance cellular uptake.172,173 The amide conjugate of NAC and cysteamine (i.e., NACMEAA) was designed to penetrate the blood–brain–barrier for applications in AD, PD, and ALS. 174 Alternatively, edits to the N-acetyl group can potentiate the activity of NAC. For example, bucillamine retains the pro-GSH activity of NAC, but is ∼16-fold more potent as a thiol donor.175–178 As a result, it can more readily scavenge ROS and has been applied clinically for both rheumatoid arthritis and COVID-19. 15 Modifications to thiol group have also been examined. For example, S-acetylation was exploited in the development of aggrelyte-2. This bifunctional NAC variant has been shown to cleave disulfide bonds and simultaneously reduce protein aggregation and AGE formation via acetyl transfer to protein lysine residues.179,180 Several additional thiol therapeutics based upon or closely related to CSH and GSH are listed in Table 2.
Lipoic acid
The cyclic disulfide lipoic acid (LA) is an endogenous cofactor produced in mitochondria of eukaryotes. Much like CSH, interest in LA as a potential therapeutic derives from its antioxidant and metal chelation properties. 181 The choline ester prodrug of LA (i.e., LACE) was shown to increase lens elasticity and advanced to human clinical trials as therapy for presbyopia. 79 However, in contrast to thiols discussed so far, LA relies on reductase enzymes to generate dihydrolipoic acid (DHLA), a reactive dithiol that participates both in redox reactions and cysteine modifications. In the context of presbyopia, it has been postulated that DHLA acts directly to cleave aberrant disulfides in crystallin protein, thereby alleviating the protein aggregation that contributes to the loss in visual accommodation. 79 Similarly, LA and its prodrug derivatives have shown beneficial effects in models of cardiovascular diseases, diabetes, and neurodegenerative diseases.181–183
Strategies to Improve Protective Thiols
Biogenic protective thiols (Section 3) serve an essential function. Depending on the context, they can act directly against disulfide bonds or indirectly by serving as reducing agents and/or scavengers for ROS/RNS. Their therapeutic counterparts (Section 6) exhibit similar potential across a large range of diseases. While many provide tangible benefits, few induce miraculous effects in vivo. Taking into consideration the strategies used by Nature and drug developers to optimize redox-active thiol small molecules, how could small molecule therapeutic thiols be further improved?
Pharmacokinetics
Whereas GSH is biosynthesized within cells and actively transported across cellular compartments by specialized enzymes, therapeutic thiols must find their way to tissue of interest. Thus, investigational compounds with poor bioavailability and/or rapid clearance may have limited efficacy in preventing or reversing disulfide formation in vivo. Additionally, the distribution of therapeutic agents is crucial, as the accumulation of stochastic disulfides varies across different organs and tissues. Given the central role of aberrant disulfides in amyloidosis, penetration over the blood–brain–barrier will be a prerequisite for therapies targeting neurodegenerative diseases. 184 So far, this challenge has been addressed by enhancing compound lipophilicity. For example, the charged carboxylate of NAC was replaced with amides (i.e., NACA and NACMEAA) to facilitate CNS-penetration. 185 Accordingly, NACA exerts superior protective effects to NAC in several models of AD and PD.186,187 Conversely, the carboxylate of LA was converted to a cationic choline ester derivative in LACE to enhance penetration into lens tissue for applications in presbyopia. 79
Prodrug design
Investigational entities bearing a free thiol functional group must contend with deleterious oxidative modifications of sulfur as they circulate. Fortunately, several effective strategies to “protect” the redox-active thiol group have been developed including the design of thioether, thioester, and disulfide prodrugs. The slow hydrolysis of thioester groups has proven to be effective, and these can be structurally tuned. For example, racecadotril features an S-acetate group that is hydrolyzed in situ to unveil the active thiol group. Conversely, erdosteine features a cyclic thiolactone that is more resistant to hydrolysis, thus slowing the rate of thiol release. 16 It can be beneficial to consider the fate of such “thiol capping” groups as well. As highlighted in the development of aggrelyte-2, the S-acetate group is transferred to lysine residues to unveil the redox-active thiolate. 180 This acyl transfer event was shown to improve protein solubility and subsequently protect lysine residues from AGE crosslinking.
Prodrug strategies may also exploit endogenous enzymes and cofactors to unveil redox-active thiol groups. For example, the cyclic disulfide in LA is unreactive and must first be reduced by GSH or oxidoreductases to generate active DHLA. As exemplified by carboxitin, disulfide prodrugs can also be used to release two distinct entities. In this case, reduction of the mixed disulfide bond releases a GSH derivative and mercaptoethylguanidine, an anti-AGE carbonyl scavenger. 146 Finally, the radioprotective agent amifostine features a unique thiophosphate group that is actively cleaved by membranous alkaline phosphatase (ALP). While ALP is abundant on the surface of healthy cells, its expression is significantly depressed in many cancer types. 188 This allows the active metabolite of amifostine (i.e., WR-1065) to selectively accumulate in healthy cells.
Thiol tuning
As highlighted by protective thiol natural products, careful tuning of the pKa and reduction potential of the thiol group is essential. More reactive thiols are not necessarily better, as reduction of native disulfides can result in protein damage. Notably, CSH-derived thiol therapeutics leverage a primary thiol group. As illustrated by GSH, MSH, and CoA, the reactivity of this group can be impacted by neighboring functionality. Changing the steric environment near the thiol group has been less systematically explored. In principle, the secondary and tertiary thiol groups of tiopronin and penicillamine, respectively, should exhibit markedly different kinetics than primary congeners. Surprisingly, aromatic thiols remain underexplored. As evidenced by thiohistidines (e.g., OSH), both the pKa and reduction potential of aromatic thiols are distinct from their alkyl counterparts.
Outlook: Implications for Longevity Science
As evidenced above, aberrant disulfides should be recognized alongside other well-established protein crosslinks (e.g., AGEs and carbamylation) as one of the hallmarks of aging. They accumulate with aging and contribute directly to the pathobiology of age-related diseases. As such, CSH-based oxidative damage to proteins fits into the context of existing aging models, including the SENS model associated with protein damage and the Hallmarks of Aging dealing with protein homeostasis. 67 The reversibility of disulfide bonds—and other oxidative modifications to CSH thiols—offers a unique avenue for the design of new strategies to combat age-related protein damage.
Despite considerable promise, this emerging field also faces challenges. Chief among them is the fact that native disulfides are part of normal protein structure and function. They are often highly conserved within proteins and tightly integrated into oxidative signaling pathways. 19 Thus, targeting aberrant disulfides selectively is difficult. Given their reversibility, the accumulation of stochastic disulfides will also be challenging to measure against the backdrop of native disulfide bonds. 189 Thus, the continued development of biochemical approaches to identify aberrant disulfides will be paramount.190,191 In addition, while stochastic disulfide formation is promoted by oxidative stress, the contributing ROS/RNS species are varied and some of them are integrated into cell signaling pathways. 192 Notwithstanding these hurdles, aberrant disulfides are distinct from AGEs. These more recognized forms of protein damage occur primarily at arginine and lysine residues and represent thermodynamic endpoints of glycation. In contrast, disulfide crosslinks can be viewed as more of an equilibrium, and thus, they are more amenable to small molecule therapy. In this regard, existing endogenous protective thiols and their therapeutic counterparts offer compelling proof-of-concept.
Future research should focus on elucidating the specific roles of CSH-derived crosslinks in age-related diseases and the effects of small-molecule thiol interventions on these parameters. New strategies to reverse damage at CSH residues are needed, and improvements in therapeutic thiols, either via iterative improvement of known thiol compounds or the design of novel chemotypes, will continue to be necessary. Further progress in this area promises to elevate protective thiols like NAC from the realm of supplements to that of approved therapeutics for aging diseases. Certainly, for achieving optimal rejuvenation in the “gotta-catch-em-all” world of age-related protein damage, the most crosslink-prone amino acid cannot be ignored.
Declaration of Competing Interest
The authors declare the following competing interests: K.M.B. and J.H.F are equity holders in LentoBio, a biopharmaceutical company focused on longevity research. This arrangement has been reviewed and approved by Florida State University in accordance with its conflicts of interest policies.