
Abstract
Charcot-Marie-Tooth disease 1A (CMT1A) is the most common autosomal dominant demyelinating sensorimotor polyneuropathy. A few patients with Charcot-Marie-Tooth disease were reported in the literature to have epilepsy. We report on an African-American boy with CMT1A, with duplication of peripheral myelin protein 22 gene, who also developed intractable generalized tonic-clonic seizures and audiovisual hallucinations.
INTRODUCTION
Charcot-Marie-Tooth disease type 1 (CMT1) is an autosomal dominant demyelinating sensorimotor polyneuropathy, previously also known as peroneal muscular atrophy. 1 Charcot-Marie-Tooth disease 1A (CMT1A) is associated with duplication of peripheral myelin protein 22 (PMP22) gene and represents 70–80% of all CMT1. 1 The prevalence of CMT1 is 15 per 100,000 and that of CMT1A is 10 per 100,000. 2 The prevalence of epilepsy in children has been estimated to be 3–6 per 1000. 3 We report on an African-American boy with CMT1A with PMP22 gene duplication, who also suffers from intractable epilepsy and audiovisual hallucinations.
CASE REPORT
This African-American boy, currently 11-years-old, was born at term after normal pregnancy, labor and delivery, and was normal until 16 months of age when he developed epilepsy with recurrent unprovoked generalized tonic-clonic seizures. He had generalized spike and wave discharges with a bifrontal predominance on electroencephalograms (Figure 1), normal blood chemistry and normal brain MRI imaging. The generalized tonic-clonic seizures were difficult to control with maximal tolerable doses of anticonvulsants, including carbamazepine with trough level up to 10 ug/mL, topiramate 10 mg/kg/day, lamotrigine 12 mg/kg/day, valproate with trough level up to 95 ug/mL and vagus nerve stimulation. He continued to have generalized tonic-clonic seizures several times a week over the last 5 years.
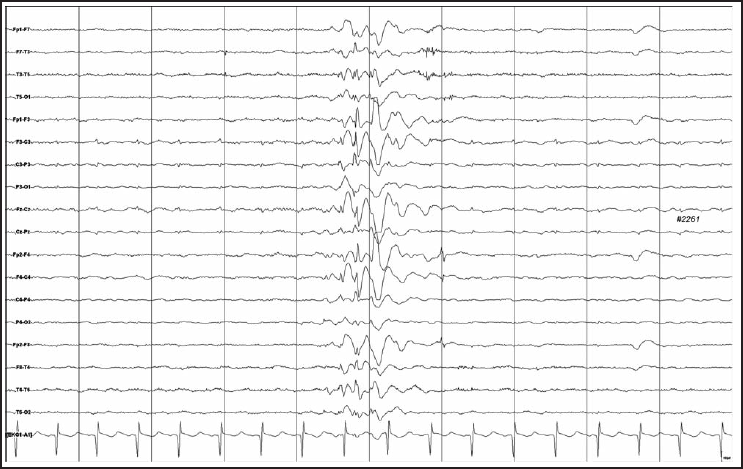
Generalized spike and wave discharges with a bifrontal predominance
The patient developed intermittent numbness, tingling and swelling of his hands and feet starting at age 6 years. Distal muscle weakness (Medical Research Council scale muscle strength: bilateral wrist extensors 2/5, bilateral ankle dorsiflexors 2/5, bilateral ankle evertors 2/5, bilateral hip flexors 4/5), generalized areflexia as well as decreased position and vibration sensation in his feet were demonstrated; otherwise physical and neurologic examinations were unremarkable. Electromyograms and nerve conduction velocity studies were performed, revealing generalized demyelinating sensorimotor polyneuropathy. He had duplication of the PMP22 gene, consistent with CMT1A. Over the last 3 years he has continued to have more severe distal weakness of all four extremities and requires a power wheelchair for long distance ambulation. He has also developed auditory and visual hallucinations and paranoid ideation, and has been treated by a child psychiatrist over the last 3 years, since age 8.
Family history reveals CMT1A in the maternal grandmother, a maternal aunt and another maternal uncle. Generalized and partial epilepsy is reported in three paternal uncles but without CMT1A. Schizophrenia is also present in the paternal family.
DISCUSSION
CMT1A is characterized by distal muscle weakness and atrophy, usually beginning in the feet and ankles; sensory loss in position, vibration, pain and temperature; slow nerve conduction velocity; and autosomal dominant inheritance. 1,2
CMT1A with PMP22 gene duplication and myoclonic epilepsy in an 8-year-old boy was recently reported. 4 This boy had CMT1A associated with myoclonic seizures and developmental delay. He was treated with carbamazepine, valproate, and lamotrigine, then seizure-free for over 2 years, and successfully weaned off his medications. No history of epilepsy was reported in the family; however, CMT1A was also seen in a brother. 4 In another report, peroneal muscular atrophy (currently known as Charcot-Marie-Tooth disease), epilepsy, cerebellar ataxia and choreoathetosis in the family were reported. 5 Seven members in three generations of the family were affected with CMT and five had CMT in combination with epilepsy. No PMP22 gene mutations were performed in the study. 5 Familial myoclonic epilepsy, ataxia, neuropathy with additional features of Friedreich's ataxia and peroneal muscular atrophy were seen in another family. 6 The mother and three of her five children had myoclonic epilepsy; the mother and one son were also ataxic; another son had additional features of Friedreich's ataxia, and a daughter had peroneal muscular atrophy. No PMP22 gene mutations were performed in the family.
At age 16 months our patient developed epilepsy, with generalized tonic-clonic seizures, which were difficult to control with maximal tolerable doses of multiple anticonvulsants including valproate, lamotrigine, topiramate and carbamazepine. Even vagus nerve stimulation was tried without benefit. The family and patient declined to try ketogenic diet for seizure treatment. At age 6 years he also complained of intermittent tingling, numbness and weakness in his hands and feet. Electromyography and nerve conduction velocity studies revealed generalized demyelinating sensorimotor polyneuropathy. PMP22 gene duplication was detected, establishing CMT1A diagnosis. His maternal grandmother, a maternal aunt and one maternal uncle were also diagnosed with CMT1A but without epilepsy. His paternal uncles suffered from partial and generalized epilepsy but without CMT1A. The epilepsy was present in our patient's paternal family and CMT1A was seen in the maternal family. The combination of epilepsy with generalized tonic-clonic seizures and CMT1A with PMP22 gene duplication in our patient is similar to the recent report in an 8-year-old boy with myoclonic epilepsy and CMT1A with PMP22 gene duplication. 4 However, epilepsy was not seen in the other members of this family. The earlier two reports did not have PMP22 gene mutation studies, however, the combination of CMT and myoclonic epilepsy was also present in these two families. 5,6 Not all patients with CMT and epilepsy have myoclonic seizures. Generalized tonic-clonic seizures but not myoclonic seizures were seen in our patient, and in another report myoclonic epilepsy was not mentioned. 5
CMT1A with central nervous system involvement such as multiple sclerosis or focal demyelization was previously reported. 7,8 Additionally, the PMP22 gene is not only expressed in the peripheral nerves, but also in the brain during different developmental stages and adult brain. 9 Whether epilepsy and CMT1A are manifestations of PMP22 gene mutations, just epiphenomenon, or due to other environmental factors is unknown and requires further studies. Since the prevalence of CMT1A is 10 per 100,000, 2 the prevalence of epilepsy in children is 3–6 per 1000, 3 the combination of CMT1A and epilepsy in this boy is extremely rare. However, he has CMT1A in his maternal family and epilepsy as well as schizophrenia in his paternal family; he may inherit CMT1A, audio-visual hallucinations and epilepsy from both sides and suffers from all these disorders.
In summary, we report a very rare combination of intractable generalized epilepsy, CMT1A with PMP22 duplication and audio-visual hallucinations in an African-American boy.
DISCLOSURE AND CONFLICT OF INTEREST
Chang Y. Tsao has no conflicts of interest in relation to this article.