
Abstract
Sporadic Creutzfeldt–Jakob disease (sCJD) is a rare transmissible disease. According to molecular classification, six clinical phenotypes of sCJD have been described: MM1, MM2, MV1, MV2, VV1 and VV2. MV2 subtype comprises 9% of sCJD cases. Atypical clinical course has been reported to be the main caveat for the diagnosis of the MV2 subtype. We hereby present a rare case of MV2 subtype of sCJD, highlighting the evolution of clinical, EEG and imaging attributes over a two-year period, thus underlining the atypical course of the disease.
Introduction
Sporadic Creutzfeldt–Jakob disease (sCJD) is a rare transmissible disease characterized by the accumulation of pathological prion protein (PrPSc) in the CNS. The polymorphism at codon 129 of the prion protein gene (PRNP) and the pattern of prion protein migration on Western blot types 1 and 2, provide a basis for the molecular classification of sCJD. 1 The MRI findings may vary accross clinical syndromes and molecular subtypes.
The typical EEG pattern of Periodic Sharp Wave Complexes (PSWC) consists of either simple sharp waves (including biphasic and triphasic waves) or complexes of spikes, polyspikes and slower waves with a duration of 100–600 ms, recurring every 0.5–2 s with amplitude of 150-300 mV and localization usually generalized, but lateralized or regional is also possible. The intervening background usually consists of generalized low voltage slowing. 2
The MV2 subtype comprises approximately 9% of sCJD cases. The atypical clinical course and the low sensitivity of established diagnostic tests have been reported to be the main caveats to the diagnosis of the MV2 subtype. 3 We hereby present a rare case with the MV2 subtype of sCJD, aiming to the evolution of clinical, EEG and imaging attributes over a two-year course.
Case Report
A 61-year-old woman was admitted to the neurology department due to evolving, over the last 4 months, cognitive decline, mild gait instability and lack of strength in the right arm and leg. The personal history was unremarkable with the exception of osteoporosis.
Neurological examination revealed limb and truncal ataxia, pyramidal involvement (Babinski sign, spasticity), apathy and gait apraxia. The patient scored 73/100 on the Addenbrooke's Cognitive Examination - Revised (ACE-R) with deficits mainly in the visuospatial and memory domains. Laboratory workup, including routine hematology, immunological, serological analysis and antibodies for autoimmune and paraneoplastic encephalopathy were unremarkable. The cerebro-spinal fluid (CSF) analysis was also within normal range. However, the Western blot assay for 14-3-3 protein was positive.
The first brain MRI, conducted 2 months since the onset of symptoms, revealed high FLAIR and DWI signal abnormalities in the right temporal, parietal and occipital cortex (Figure 1A-B-C-D). Brain MR angio- and venography (MRA-MRV), cervical MRI and total-body CT were unremarkable. The EEG conducted 4 months since symptom onset (Figure 2A1, A2) revealed lateralized synchronous semi periodic, sharp wave complexes of triphasic morphology 3.5-4 Hz, at the central - posterior regions mainly of the right hemisphere and background rhythm of 9 Hz at the occipital regions.
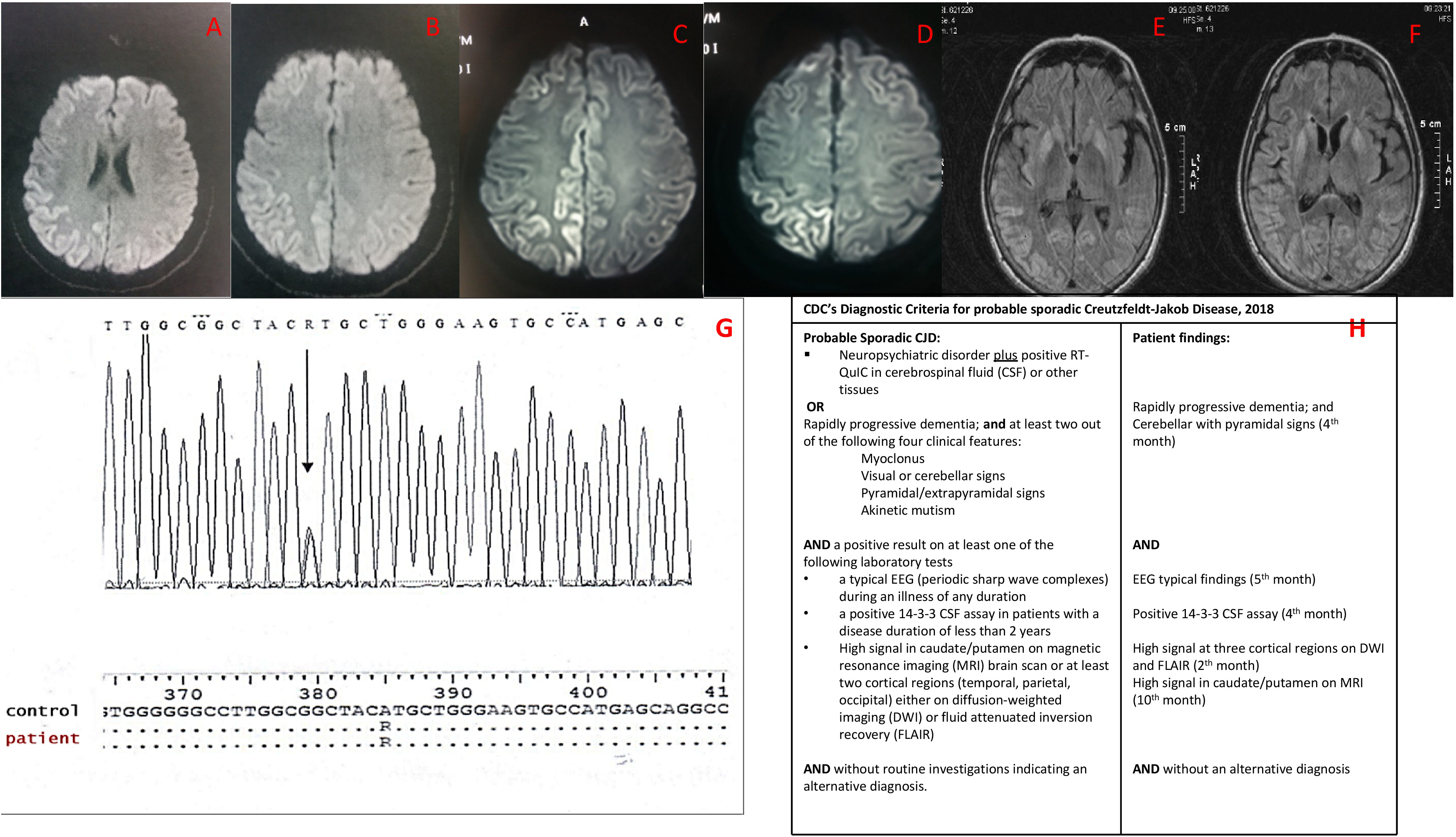
A – F: MRI evolution of sCJD MV2 subtype, G – H: RPNP sequencing and CDC's diagnostic criteria for probable sCJD. A, B, C, D. second month: A-B: MRI-FLAIR: Hyperintensities in the right parietotemporal cortex and the occipital cortex (cortical ribbon sign). C-D: MRI-DWI: Hyperintensities in the right temporal cortex and the occipital cortex (cortical ribbon sign). E, F. 10th month: MRI-FLAIR: bilateral, asymmetric, cortical hyperintensities and bilateral striatal hyperintensities. G. RPNP sequencing: Shows the point mutation (Alanine to Arginine) at codon 129 of the PRNP gene, causing the expression of Valine instead of Methionine. H. CDC's Diagnostic Criteria for probable sCJD.
At this time-point, based on the clinical features (subacute progressive dementia, pyramidal and cerebellar signs), the MRI findings (cortical ribbon sign) and following the exclusion of other alternative diagnoses, the patient was diagnosed with probable Creutzfeldt-Jakob disease (sporadic form), according to the recently revised diagnostic criteria of the Centers for Disease Control (CDC) (Figure 1H). 4
The patient was followed thereafter on an outpatient setting. Serial EEGs were as follows: 5 months since the onset of symptoms evident generalized PSWC of high voltage (120-150 uv) triphasic sharp waves and complexes of spikes-waves, lasting 240 - 450 ms, every 1–3 sec, mainly at the central-posterior regions with right lateralization. The intervening background consisted of generalized low voltage mild slowing of 8.5 Hz (Figure 2B1, B2). 6 months since the onset of symptoms, evident semi periodic sharp waves of lower voltage mainly at the posterior regions with a background rhythm of 9 Hz at occipital regions. (Figure 2C).

EEG evolution of sCJD MV2 subtype (Machine settings display: sensitivity 10 μv/mm, time constant (s) o.3s, high frequency filter 70Hz, paper speed 10 sec per page/screen).
The patient was newly admitted ten months after the onset of symptoms due to clinical deterioration. She was presented with rapidly progressive dementia (53/100 on ACE-R), myoclonus, pyramidal signs, apraxia in four extremities, extrapyramidal signs (rigidity, tremor), and severe gait disorder (astasia). The patient did not experience clinical or electrographic seizures during the course of the disease.
The EEG on 10th month, revealed diffuse slowness of 6 Hz at the posterior regions and continuous recording of slow polymorphic waves of 2-3 Hz at the frontotemporal regions, intermixed with bilateral waves of triphasic morphology. Compared to previous recordings there was diffuse slowness with atypical paroxysmal findings (Figure 2D).
Brain MRI on 10th month from the onset of symptoms, revealed bilateral, asymmetric, cortical hyperintensities and bilateral, striatal hyperintensities on T2, FLAIR and DWI, with restricted diffusion on ADC map (Figure 1E-F).
Genetic testing (sequence of PRNP gene) did not reveal a previously described mutation linked with familial CJD, except for the heterozygous substitution of methionine from valine at codon 129 of the PRNP. (Figure 1G).
The identification of the MV2 molecular subtype assisted with the diagnosis. On a 12-month follow-up, the patient received symptomatic treatment with clonazepam and levetiracetam at low doses. The patient was bedridden and gradually exhibited akinetic mutism resulting to death due to a respiratory infection.
Discussion
We hereby present a female patient with sporadic CJD. Although at the time of admission sporadic CJD was highly likely diagnosis, a wide variety of alternate diagnoses required investigation in order to be excluded, such as Hashimoto's encephalitis, Wernicke's encephalopathy, toxic/metabolic encephalopathy, infective encephalitis, non-convulsive status epilepticus, brain tumor or metastases and vascular causes. Thorough disease history, extensive laboratory evaluation, neuroimaging and EEG findings were essential for the diagnosis.
The most important CJD mimics, namely, antibody-mediated inflammatory disorders of the central nervous system, including autoimmune encephalitis, were excluded based on the unremarkable CSF analysis and the absence of autoantibodies linked with these pathologies.
Then the differential diagnosis was essentially limited to neurodegenerative diseases, namely, Alzheimer disease, dementia with Lewy bodies and tauopathies. However, these conditions are characterized by a prolonged course of deterioration, compared to the presented patient. Moreover, the presented patient did not exhibit the distinct clinical characteristics and MRI imaging attributes observed in the context of these neurodegenerative diseases.
The patient fulfilled the currently revised CDC's diagnostic criteria for probable CJD (Figure 1H) 4 by the fifth month of the disease, based on the disease history, the subacute cognitive deterioration, the cerebellar and pyramidal signs, the MRI and EEG findings and the presence of 14-3-3 protein in CSF.
Of note, four months after the symptoms’ onset the patient exhibited atypical EEG pattern, whereas in the majority of CJD cases, diagnostic EEG findings reportedly appear at approximately 12 weeks following the disease onset. Due to this observation, the diagnosis was especially challenging. Also, the evolution of the EEG recording 10 months after the symptoms’ onset into encephalopathic pattern without findings typical of CJD poses additional challenges with respect to disease follow-up. PSWC typically disappear in later stages of sCJD and the EEG at these stages presents with low voltage activity followed by electrocerebral inactivity. 3
With respect to the clinical presentation, although the patient manifested early apathy, and apraxia, myoclonus, a typically early symptom in sCJD, only presented 7 months after the disease onset. 5
The evolution of MRI presentation included early invasion of the cortex from the second month of the disease by later invasion of the basal ganglia. This evolution of MRI findings is the only typical finding of sCJD in the presented patient. The median time between the appearance of cortical ribbon sign and basal ganglia lesions in patients with sCJD is 2.7 months. 6
According to the genetic analysis, including PRNP codon 129 polymorphisms, our patient was found to be methionine/valine heterozygote. This polymorphism affects not only the clinical expression but also the duration of the disease. MV2 clinical phenotype presents with a longer duration (mean 17,1 months), atypical PSWC in EEG,3,7 progressive dementia, ataxia and neuropsychiatric features such as behavioral abnormalities, aphasia, apraxia, and frontal lobe syndromes.
An effect of prion protein type on MRI presentation has not been reported to date, although there is a reported tendency for higher MRI sensitivity in terms of diagnostic accuracy in patients with prion type 2 protein. The EEG and the CSF examination for 14-3-3 protein exhibit higher diagnostic sensitivity in the prion protein type 1 disease. 3 These findings correspond to the characteristics of our patient.
Our study describes the evolution of EEG and MRI, as well as the clinical features over 10- month follow-up of patient with MV2 sCJD. Specifically, the appearance of the typical EEG pattern was observed in the fifth month of the disease course, whereas myoclonus was detected for the first time 6 months into the disease course.
At 10 months, the EEG transitioned to an atypical moderate encephalopathic pattern in the absence of severe encephalopathic pattern, characterized by low voltage or electrocerebral inactivity. With respect to the MRI evolution, the patient presented typical MRI for sCJD in the second month of the disease with hyperintensities in three cortical areas, followed by the development of asymmetric bilateral cortical invasion with simultaneous involvement of the basal ganglia.
In conclusion, the diagnosis of sCJD MV2 subtype cases poses special challenge due to atypical characteristics. We hereby present a case of a patient exhibiting the evolution of EEG and imaging findings in the context of sCJD MV2 subtype with a long survival period of two years. Our study, describes atypical EEG features, clinical signs and symptoms, as well as imaging findings during the course of the disease, characteristics that may be helpful for the Neurologist towards the accurate diagnosis of patients with sCJD MV2 subtype.
Footnotes
Authors’ Contributions
AT performed EEG evaluation, data collection, conceptualization and writing of the manuscript. SS-A, IP, ST, BM collected data, reviewed and contributed to the manuscript. GN critically reviewed the manuscript. All authors read and approved the final manuscript.
Research Ethics and Patient Consent
Written consent after being informed of the publication of patient information and images was given by a legally authorized representative.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Not applicable, because this article does not contain any studies with human or animal subjects.