
Abstract
Impracticability is an ethical standard for waiver of informed consent in research. We examine how well the criterion of impracticability appears to have been fulfilled in a set of 36 completed randomized controlled trials (RCTs) that secured consent from some subjects or LARs and employed waivers to enroll others. These trials were identified among 155 RCTs using waivers of consent in a convenience sample drawn from 7 systematic reviews. Recruitment data were available for 19 of the 36 trials, revealing an average of 41.6% of subjects (range 0.2–98.7%, 95% CI: 24.8–58.4%) were enrolled without consent. Six trials enrolled less than 10% of subjects without consent and an overlapping set of 9 trials sought consent from all subjects or LARs at some sites while waiving consent at other sites. We question whether these trials were practicable without waivers and identify issues for consideration by investigators and ethics review boards.
Keywords
Informed consent is a cornerstone requirement of human subjects research, particularly in randomized controlled trials (RCTs) (Benatar, 2002; Miller, 2014; Capron, 2018). Nonetheless, research ethics guidelines and national laws have recognized certain exceptions to this requirement. The standards for performing research without consent vary widely, though there are some common elements (van Belle et al., 2015; Rebers et al., 2016; Flory et al., 2016). For example, trials that may be performed without consent include those that pose no more than minimal risks to subjects, involve only treatments accepted as standard practice, are cluster randomized, are considered pragmatic or real-world, or include some combination of these features (Flory et al., 2016; Zhang et al., 2021). Many countries allow trial investigators to forgo informed consent when testing interventions for emergency conditions (e.g., cardiac arrest). In these cases, patients are often incapacitated, and the therapeutic time window may be too narrow for investigators to seek consent from a legally authorized representative (LAR) (Rebers et al., 2016; Flory et al., 2016; World Medical Association, 2013a). Rules vary, however, on what procedural safeguards must be in place, and whether patients must be notified or asked for consent when they regain capacity.
A necessary but not sufficient condition for permitting research without consent is that consent cannot be secured or would be impracticable or the act of seeking consent would render the research impracticable (Council for International Organizations of Medical Sciences, 2016a; National Health and Medical Research Council and Australian Research Council, 2007, 2018; U.S. Department of Health and Human Services, 2019a; U.S. Department of Health and Human Services, 2019b; U.S. Food and Drug Administration, 2018; Tri-Council, 2010, 2014; New Zealand National Ethics Advisory Committee, 2019). We understand the meaning and application of these standards to be equivalent. In our view, the practicability of securing consent is a characteristic of the population to be studied. Thus, if a large majority of a target population is foreseeably capable of consent, then securing consent should be judged to be practicable.
Even though the requirement of impracticability is central to the justification for foregoing informed consent, there is little empirical data providing insights into how well the requirement has been fulfilled in RCTs. A recent review by Zhang and colleagues found that 165 trials out of their full sample of 1988 pragmatic RCTs waived consent for at least part of the trial procedures. They examined whether investigators offered justifications for the waiver, and 76 (46.1%) papers provided a total of 118 justifications. Nineteen justifications appear relevant to the impracticability standard, including 10 where securing consent would make the study “impossible or infeasible,” 5 where “consent would bias the response or the results,” and 4 “emergency research” (Zhang et al., 2021).
Notably, the impracticability issue was raised after the first trial performed (in part) under the U.S.'s 1996 Exception from Informed Consent (EFIC) rule, the National Acute Brain Injury Study: Hypothermia trial. The trial was run for 9 months with consent from LARs; however, because of low recruitment and delayed enrollment, the study was going to be terminated. Subsequently, a waiver of consent was granted for subjects for whom LAR permission could not be secured within 1.5 h after hospital admission. The use of waiver was criticized because 62% of subjects were enrolled with consent. The investigators performed a detailed analysis of enrollment, finding that minorities were underrepresented during the first 9 months due to difficulties contacting LARs and not due to refusals (Clifton, personal communication, 2021). The disparate waiving of consent for minority subjects was justified on grounds of ensuring equitable inclusion and generalizability of the trial's results (Clifton et al. 2002).
We are performing a long-term project collecting information about RCTs involving waivers of informed consent. We recognized that some trials secured prospective consent from some subjects or their LARs but waived consent from others. For example, the Acute Respiratory Distress Syndrome Network ARMA trial, which randomized ICU patients on mechanical ventilators to different tidal volumes, waived consent at one of 10 trial sites, and 16 of 861 (1.8%) subjects were enrolled without prospective consent. While the waiver was justified to allow inclusion of emergency patients at the one site (The Acute Respiratory Distress Syndrome Network, 2000; Baker & Merz, 2018), inclusion of those subjects was unnecessary for completion of the trial. Another trial, the Nice-Sugar trial, compared tight versus loose glucose control among critically ill patients. The trial was conducted with delayed consent at the 67 sites in Australia and New Zealand and with prospective consent at all 17 sites in Canada (The NICE-SUGAR Study Investigators, 2009).
These examples, in which large numbers of subjects were enrolled with consent, challenged our conception of the bounds of impracticability as a necessary condition for performing research without consent. Thus, we performed an interim review to better understand and draw lessons from the nonexclusive use of waivers of informed consent in clinical trials.
Methods
As a general matter, we consider any trial that does not secure prospective informed consent (written or verbal) based on a “full” disclosure (i.e., providing sufficient informational content to fulfill local ethical and regulatory standards) from all subjects or their LARs or family members to involve a waiver. We use the term “waiver” regardless of the language used by investigators to describe their processes, including waiver or modification of informed consent (WIC), opt-out as permitted under the US Food and Drug Administration's Exception from Informed Consent (EFIC) rule (U.S. Food and Drug Administration, 2018), and “deferred,” “delayed,” or “retrospective” consent. From a regulatory perspective, consent processes that provided brief, short-form, abridged, or scripted disclosures, or otherwise withhold information (e.g., about randomization or placebo controls) prior to randomization or enrollment may also be considered waivers, because the normal expectations for prospective informed consent (as defined above) are set aside. For the purposes of this study, however, we focus more narrowly on trials that have waived any form of prospective disclosure of information about the trial that would provide potential subjects or their LARs an opportunity to object prior to randomization and intervention.
In the course of performing other studies over the last 5 years, we identified systematic reviews of trials that include detailed information about or are specifically focused on the use of waivers of informed consent. When we identify relevant reviews, we secure lists of included trials from investigators or supplemental materials. Here, we present an interim analysis of trials drawn from 7 recent reviews. This is a convenience sample drawing on our own work as well as several other reviews.
Feldman and colleagues examined 36 trials published in 2010 through mid-2014 testing acute interventions for ischemic stroke (Feldman et al., 2016). Armstrong and colleagues identified 53 papers (for 45 trials conducted between 2000 and 2016) involving pre-hospital recruitment of patients (2017). A more recent study by Feldman, Hey & Kesselheim identified 41 trials approved since 1996 under the FDA's EFIC rule, presenting detailed information on 33 trials completed at the time of their review (2018). We updated the trials that have since been completed and published. Klein, Moore & Biros performed a systematic review of 20 years of published trials using EFIC or WIC, identifying 28 trials (2018). Taljaard and colleagues examined a random sample of 40 papers (39 trials) using cluster randomization for individual-level interventions and published between 2007 and 2016 (2020). Dhamanaskar & Merz performed a systematic review examining the 100 most-highly-cited RCTs sampled from Scopus each year for 2014–2018, identifying 44 trials (8.8%) in which fully-informed prospective consent was not secured (2020). Finally, Lin, Jochym & Merz performed a systematic review of investigator-described pragmatic or comparative effectiveness RCTs published in 2014 and 2017 with one or more US sites, finding 24 RCTs (23% of their sample) that waived informed consent, including one trial that withheld information about randomization (2021). We removed duplicate trials, leaving a core set of 215 trials dating back to the late 1990s.
The 215 trials were all summarized, and supporting protocols and supplemental materials were examined if necessary to understand the trials. For studies that lacked recruitment details, we contacted authors by email or phone in order to describe consent methods and recruitment results. Querying authors for details about their trials is considered non-human subjects research by the University of Pennsylvania IRB.
We identified trials that involved waivers of informed consent, and then we focused more narrowly on the subsample of trials in which consent was sought from some subjects but not others. Qualitative analyses were planned, as well as contingency table and nonparametric quantitative assessment of rates of recruitment without consent and, for emergency research, therapeutic time windows for study intervention (Stata 12.1, StataCorp, © 2014). There was no funding for this study.
Results
From the core of 215 unique trials identified from the 7 source reviews, we were able to gather information about consent for 213 trials. Of these 213, 155 trials waived consent for at least some subjects. One hundred and one (65.2%) of these trials had sites in the USA, Canada, or Australasia, where rules explicitly require a judgment of impracticability (in New Zealand, that consent “cannot” be secured) for consent to be waived (hereinafter the “impracticability” countries). One hundred and two of these trials (65.8%) tested emergency interventions, and at least 62 of these (2 are missing data) involved prehospital interventions, reflecting the sampling frames of the papers from which we drew our sample. Thirty-six (23.2%) of the 155 trials secured consent from some subjects and waived consent from others. These 36 trials are the primary focus of this analysis. Each trial is described in Table 1, with a summary of available recruitment data.
Enrollment Methods and Results in Trials Securing Consent from Some Subjects and Waiving Consent from Others.

Notes: †: Percentage of subjects enrolled without any notice or opportunity of the subject or their LAR to refuse or opt-out.
: Data provided by authors.
: Data collected by Feldman et al. (2018).
NR: Not reported.
NA: Not available.
USA: United States.
EFIC: US Food and Drug Administration Exception from Informed Consent regulation (2018).
Published papers on 9 of the 36 trials provided full recruitment details, and we contacted authors from the remaining 27 trials in order to solicit recruitment data or missing details. We received useful responses from 20 (74.1%). We have no recruitment information of any kind for 4 trials. Respondent authors of 8 trials indicated that they had not collected, coded, or analyzed recruitment data. We have limited information about a further 5 trials, including: 1) three multisite trials, 2 in the US and 1 in Australia and New Zealand, required consent at some sites while granting waivers at others, but recruitment data are unavailable; 2) a trial in the Netherlands and Germany secured written consent from subjects in the ambulance prior to randomization and treatment, with a “minority” providing consent after arrival at the hospital; and 3) a trial in Ireland provided data on the number of subjects who consented for themselves and the number of refusals (all by surrogates), but collected no further details about the extent to which the treatment team involved family members in the recruitment decisions to enroll incapable patients. Thus, we determined rates of recruitment with waivers of consent for 19 trials: data were provided in 7 papers, collected by Feldman et al. (2018) for 4 trials, and provided to us by trial investigators for 8 trials.
The percentage of subjects recruited without any form of prior notification and consent ranged from 0.2 to 98.7% (mean = 41.6, 95% CI: 24.8–58.4). Six trials enrolled more than 90% of subjects with patient or family consent, 3 of which secured consent from all subjects at some sites but waived consent for at least some subjects at other sites. Overall, 9 trials (including 5 for which we have no recruitment data) secured consent at some sites while waiving consent at others; this typically reflected different regulatory or IRB/REB requirements. These data raise the question of why the trials were considered to be impracticable unless the prospective consent requirement were waived.
Eighteen of 36 trials in the full set (50%) and 12 of 19 (63.2%) trials for which we have recruitment data were performed in the impracticability countries. Two additional multinational trials included sites in the US, Canada, and Europe, but waivers were used only at sites in Europe. The average percentage of subjects recruited without consent in the 12 trials conducted in impracticability countries (49.2%) appeared somewhat higher than the average percentage recruited in trials run elsewhere (28.7%), but the difference was not significant (p = 0.20 by nonparametric Mann-Whitney test). Forty-two of 155 trials (27.1%) that waived consent were cluster-randomized, but only 3 of these 42 (7.1%, Fisher's exact p = 0.003) also sought consent from some subjects, reflecting the prevalence of waiving consent from all subjects in cluster-randomized designs.
Thirty-one of the 36 trials (86.1%) and 17 of 19 (89.5%) trials for which we have recruitment data involved emergencies. Of the 31, 11 trials tested interventions for stroke, 6 for traumatic brain injury, and 5 for other trauma, hemorrhage, or shock. As shown in Table 2, waivers were very likely to be used in trials for treatment of cardiac arrest, traumatic brain injury and other trauma, hemorrhagic shock, respiratory distress, and critical illness not otherwise specified. Overall, consent was sought from at least some subjects or their LARs in the overwhelming majority of trials (37 of 41, with 2 trials indeterminate) in stroke.
Prevalence of Waivers of Consent by Diseases or Conditions Involved in Trials.
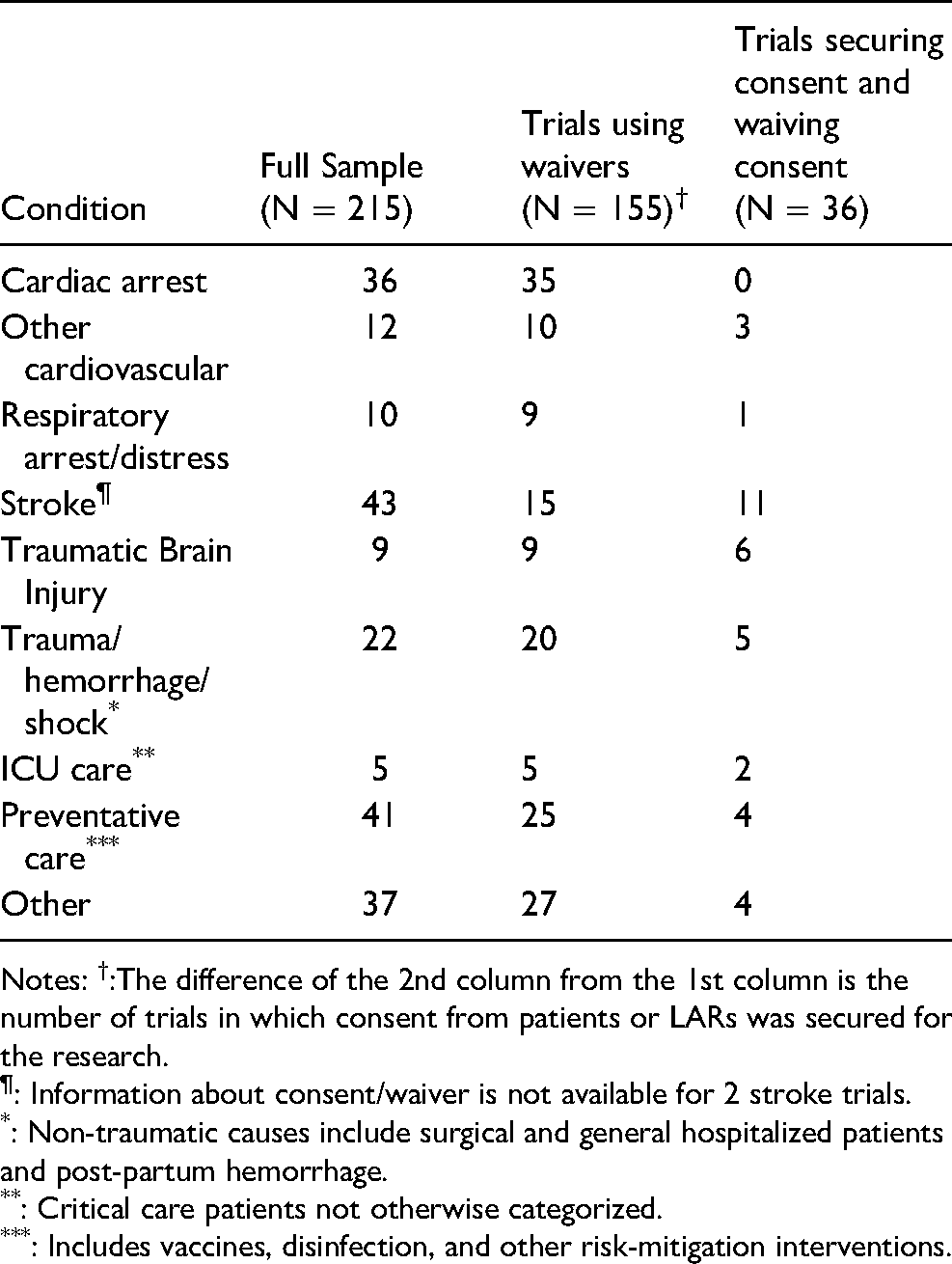
Notes: †:The difference of the 2nd column from the 1st column is the number of trials in which consent from patients or LARs was secured for the research.
: Information about consent/waiver is not available for 2 stroke trials.
: Non-traumatic causes include surgical and general hospitalized patients and post-partum hemorrhage.
: Critical care patients not otherwise categorized.
: Includes vaccines, disinfection, and other risk-mitigation interventions.
To examine whether emergency trials which allowed for greater therapeutic time windows were more likely to seek consent, we coded inclusion and exclusion time windows when available in published reports or protocols (when readily available). We assumed immediacy for cardiac arrest and pre-hospital interventions unless authors indicated otherwise. Time window definitions varied widely (e.g., time from symptom onset or injury, time from hospital arrival, time from diagnostic test, and time from emergency dispatch call), limiting comparison across trials. Of course, a time window is not meant to imply that trial interventions are not initiated as quickly as possible; we thus interpreted these windows to be a crude index of urgency. We coded time windows for 124 trials involving emergent care, 70 of which waived consent from all subjects. Twenty-six trials secured consent from some subjects, and 28 trials secured consent from all subjects. Of the 70 trials fully waiving consent, 56 (80.0%) had time windows of zero, and the remaining 14 reported windows ranging from 1–12 h (mean of 5.2 h). Of the 54 trials seeking consent when possible, 7 trials (13.0%) had time windows of zero, with the other 47 trials reporting windows ranging from 1–24 h (mean of 7.0 h). Thus, those trials securing consent when feasible had more time available than trials waiving consent from all subjects (p < 0.0001 by Mann-Whitney test).
Finally, we examined whether trials that had longer time windows enrolled a greater percentage of subjects with consent, regressing the rate of enrollment without consent by the reported time windows. We have 14 trials with this data, and the result suggests that the rate of enrollment using waivers drops as the time window increases, but this was not significant in our sample (t = -1.46, p = 0.17).
Discussion
This is the first study to examine how well the criterion of impracticability has been fulfilled in RCTs involving waivers of informed consent. We find that almost a quarter of the 155 trials in our sample using waivers of informed consent secured consent from some subjects. Of these 36 trials, we obtained recruitment data for 19 trials, finding that 42% of subjects on average were enrolled without any form of prior notification that would have enabled the subjects or their LARs to opt-out or consent. As noted above, 6 trials enrolled less than 10% of subjects without consent and an overlapping set of 9 trials sought consent from all subjects or LARs at some sites while waiving consent from at least some subjects at other sites. We believe that these findings raise a question about whether the trials could have been carried out if restricted to subjects from whom personal or LAR consent was secured; that is, whether the trials were in fact practicable without waiving consent from any subjects or their LARs.
Our data also show that, on average, emergency trials in which consent was sought from at least some subjects or their LARs reported having longer therapeutic time windows available than trials waiving consent from all subjects. We make no judgment about the appropriateness of waiver in any given trial (particularly those in which consent was waived from all subjects), but believe these findings suggest a need for closer scrutiny of the impracticability of carrying out research in which waivers are proposed.
We have used our findings to distill characteristic features of RCTs that have been performed with prospective consent from some subjects or LARs but no prior consent from others. The nonexclusive categories include: 1) different target populations (as in the ARMA trial discussed above); 2) different interventions (e.g., a trial comparing experimental treatment vs. usual care); 3) different sites (perhaps reflecting the decisions of different IRBs/RECs and different regulatory requirements); and 4) for emergency research, the therapeutic time available to seek prospective patient or LAR consent, with longer time windows increasing the likelihood of being able to ask for consent. Each justification for discriminant use of waivers raises unique issues and concerns that must be addressed in the design and ethical approval of trials. The one common thread, of course, is that, along with other regulatory requirements, the conduct of the trial must be determined to be impracticable unless the prospective consent requirement is waived.
While impracticability is an explicit regulatory requirement in at least half the trials in our sample, it reflects the important ethical standard that research should involve the least vulnerable subjects necessary in order to answer the research question (World Medical Association, 2013b; U.S. Department of Health, Education, and Welfare, 1979; New Zealand National Ethics Advisory Committee, 2019). Waiving consent, of course, renders subjects vulnerable because of their complete inability to protect their own interests when they are subjects of research about which they or their family members have no knowledge (Council for International Organizations of Medical Sciences, 2016b).
Impracticability is a high standard for foregoing informed consent. “Impracticable” is defined as “impossible to carry out” (Lexico.com, 2021). “Practicable” means “capable of being put into practice or of being done or accomplished” (Merriam-Webster, 2022). Early US regulators and the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research envisioned waivers as being limited in scope and used only when absolutely necessary to carry out critically important research, and expressed worry about waivers becoming routine or used for convenience (Kassels & Merz, 2021). The US Department of Health and Human Services Secretary's Advisory Committee for Human Research Protections (SACHRP) and other commentators have suggested that impracticability of research should be based on considerations of: 1) scientific integrity, such as avoiding selection or response biases; 2) ethical issues, such as privacy; and 3) the scientific and ethical rationale for why research cannot be performed with subjects capable of consent (2008). As SACHRP noted, impracticability should not turn exclusively on “convenience, cost, or speed” (2008), an opinion shared by many others (Gelinas et al., 2016; Kalkman et al., 2017; Murdock & Caulfield, 2018). In our opinion, the bigger problem with the regulatory threshold of impracticability is that it fails to require any assessment of the importance of the research and to balance that against the social value of informed consent (e.g., promoting respect for persons). For example, an IRB/REB may approve a study of a clinically and scientifically unimportant question to be conducted without consent because of potential bias, as long as the criteria (e.g., offering perhaps minimal potential benefit but posing no more than minimal risk to subjects) for waiver are met. This approach we believe undervalues informed consent.
Trials in specific emergency conditions suggest consent is often feasible, at least from some subjects or available LARs. For example, two-thirds of trials involving traumatic brain injuries sought consent when possible. Five of 6 trials involving patients with suspected or diagnosed ST-Elevation Myocardial Infarction (STEMI) secured some form of oral or written assent or consent from patients or LARs before randomization by emergency personnel before or during transport to a hospital (ten Berg et al. 2010; Montalescot et al., 2014; Selker et al., 2012; Steg et al., 2013; Stub et al., 2015), while one trial completely waived consent for all patients entering the catheterization laboratory (Shahzad et al. 2014; Dickert et al., 2018). Nearly every stroke trial secured consent from at least some subjects or LARs. As one investigator told us by email, waivers in stroke trials may be problematic because information about time of symptom onset may be a specific criterion for inclusion, absence of which makes a patient ineligible. Thus, if the patient is incapable and no family is available, a waiver is ineffectual.
While clearly informed by hindsight, we believe there are several lessons to learn from this review. First, investigators should clearly establish why their studies would be impracticable if they were required to enroll only subjects capable of consenting (or having LARs available to do so on their behalf), and ethics review boards should ensure those reasons are legitimate (Capron, 2018, at 23; London et al., 2020). We believe it is incumbent on clinical researchers and ethics reviewers to provide explicit justification for withholding information that would typically be disclosed to trial participants and for their decisions to “waive” usual consent requirements. For example, if a trial is designed to examine treatment efficacy across a broad range of eligibility, and the likelihood of incapacity or unavailability of LARs is foreseeably heightened for some targeted subpopulation (such as those who are more seriously injured or debilitated, where the expected sample size will be sufficient to perform planned subgroup analyses), the waiver and use of different approaches to consent may be appropriate. Disparate impacts on people of color and those with poor access to resources are foreseeable, but the use of waiver has been justified as a means for ensuring just distribution of the burdens and benefits of research (Yamal et al. 2014; Clifton et al. 2002; McCarthy, 1995; U.S. Food and Drug Administration, 1995; Sugarman et al., 2009). Nonetheless, disproportionate application of waivers to minority populations may be interpreted to be exploitative, evoking a history of abuses (Dula, 1997; Public Citizen, 2021). Moreover, using waivers to overcome low enrollment or imputed biases in enrolled populations attributable to treatment or research participation preferences is not legitimate (Truog et al. 1999; Largent et al., 2010). If investigators and ethics review boards are uncertain about the necessity for a waiver in order to perform a proposed project, they should examine experiences of recruitment in similar trials involving the same target populations. They may also adopt an experimental approach, trying different methods and assessing to what extent securing consent undermines trial performance.
Second, for multisite and multinational trials, ethics review boards should be aware of what other IRBs or RECs have done, and investigators, who are in control of IRB/REC communications, should let boards know if other boards or institutions have required different approaches to avoid substantially different consent methods across trial sites and to ensure that potential subjects are treated equitably. It strikes us as unfair for some subjects (or their LARs) to be informed and asked for their consent while other subjects are not, absent substantial justification (e.g., inclusion of different target site populations each critical for answering specific research aims for those populations but with divergent expectations for patients’ capacities for consent) for treating similarly situated persons differently. Notably, cross-site differences may become less of an issue with implementation of single IRBs of record for federally-funded RCTs performed wholly in the United States (U.S. Department of Health and Human Services, 2019c).
Third, trials typically span several years during which time investigators should collect recruitment data and IRBs and RECs should monitor trial progress for assessment of the continued necessity for the waiver. Detailed recruitment data should also be published, as greater transparency for trials involving waivers may go a long way to developing public acceptance of these research methods (Yamal et al., 2014; Clifton et al., 2002; Sugarman et al., 2009).
There are several limitations to this interim study. First, we conceptualize a very broad definition of “waiver,” subsuming a range of conceptually distinct ethical and regulatory approaches that permit enrollment of human subjects in RCTs without their individual, fully-informed prospective consent (or that of their LAR). We have focused on a subset of waivers having the common feature that subjects or their LARs are not informed or given an opportunity to refuse prior to experimental intervention. We draw no conclusions about the handful of trials in our sample that included other modifications of the consent processes. Second, we have glossed over any differences in regulatory language, including impracticability of research in which consent is required and impracticability of securing consent. Third, we have not systematically searched protocols or other trial communications to examine researchers’ arguments and justifications for waiving consent. Fourth, we did not perform a systematic review for relevant reviews or for RCTs that used both waivers and secured consent from subjects, and our sample draws heavily on trials performed with waivers in the USA and other impracticability countries. Our sample comprises a large number of RCTs using waivers of consent that is highly heterogenous. These factors limit our ability to analyze the representativeness of our sample to the greater universe of trials involving waivers, including an assessment of different regulatory standards for foregoing prospective consent, and limits our ability to dig more deeply into trial details. As mentioned, this is an interim analysis of what we hope will be a substantially larger collection of trials for analysis that will yield more insights in the future.
In conclusion, this study provides useful insights into the performance of RCTs without prospective informed consent from all subjects or their LARs, shedding light on how well the standard of impracticability has been fulfilled, and suggesting a range of considerations aimed at improving decisions of researchers and ethics review boards about the appropriate use of waivers.
Best Practices
Researchers should thoroughly assess and justify their proposed waivers of consent from subjects, and strive to ask for consent from subjects or their legally authorized representatives whenever possible.
Institutional ethics review boards should ensure that researchers have justified waivers of consent, striving to ensure that the reasons are legitimate, that securing consent from all potential subjects or their LARs would truly undermine the performance of important research, and that there be legitimate research-related justifications for treating potential subjects differently.
Research Agenda
This study highlights the ambiguity in the regulatory standards supporting research performed without informed consent from all subjects. Researchers should be clear about the critical importance of their research and the threats to completion of studies if consent is required. Institutional ethics review boards should pay close attention to trials in which waivers are requested and granted, ensuring that similarly situated people are treated equitably. Further research may help further flesh out legitimate conditions for waivers, enabling greater calibration of researchers and ethics boards in the future.
Educational Implications
The results of this study suggest the need for education of IRB members and clinical trialists about the interpretation of impracticability and the need for researchers to demonstrate that their research would be nigh impossible to perform if they were required to secure informed consent from potential subjects. IRBs should also be sensitive to monitoring multi-site approvals to assure all subjects are treated equally, and should monitor research in which waivers have been granted to assess the continued necessity of foregoing informed consent.
Footnotes
Acknowledgments
The authors thank the systematic review and trial investigators who responded to our inquiries and provided detailed information about their studies, and Lois Shepherd, Amy Miller, and several anonymous reviewers for comments. Responsibility for the study is solely that of the authors. Funding: none.
Author Contributions
JFM conceived of the idea for the study, collected recruitment details for some of the trials, and performed the quantitative analysis; WBF collected recruitment details for some of the trials, and the authors contributed equally to the coding, checking, and interpretation of data, and the editing of the manuscript. RD wrote the first version of part of the paper, and JFM wrote first versions of part of the paper and managed subsequent editing.
Competing Interests
Since 1997, JFM has been a paid expert witness in 3 civil cases involving the adequacy of informed consent and IRB review in research, twice for defense and once for plaintiff, as well as a 4th case for plaintiffs involving the definition of human subjects research. In the last 3 years, JFM has received financial compensation for service on several Data and Safety Monitoring Boards for the NIH and the American College of Radiology Imaging Network, and for service on a pharmacogenomics ethics advisory board for Merck. JFM received partial salary support as moderator of the IRBForum (![]() ) by a grant from Public Responsibility in Medicine & Research (PRIMR) from 2012 through 2020.
) by a grant from Public Responsibility in Medicine & Research (PRIMR) from 2012 through 2020.
Within the last 3 years, outside the scope of this work, WBF has served as a paid consultant for Alosa Health and Aetion. He received an honorarium for a presentation to Blue Cross/Blue Shield of Massachusetts, and is funded by an unrelated grant from the National Institutes of Health (5T32HL007633-35).
RD reports no conflict of interest.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.