
Abstract
Asthma is a heterogeneous respiratory disease with complex pathogenesis involving immune dysregulation, environmental triggers, and increasingly recognized to have contributions from the human microbiome. Emerging evidence from longitudinal birth cohorts and multi-omics studies reveals that early-life microbial colonization patterns in both the gastrointestinal and respiratory tracts play a crucial role in shaping immune trajectories and influencing asthma susceptibility. This expert review highlights the findings from pivotal studies that associate dysbiosis in the gut and airway microbiota with asthma development and its diverse phenotypic manifestations. Reduced abundance of immunomodulatory genera such as Bifidobacterium, Faecalibacterium, and Lachnospira in the gut has been consistently associated with increased asthma risk. In the airways, increased colonization by potentially pathogenic taxa, including Moraxella, Haemophilus, and Streptococcus, correlates with viral respiratory infections and persistent wheezing. Microbiome diversity patterns also differ between asthma phenotypes: eosinophilic asthma typically features a community profile closer to healthy individuals, while neutrophilic asthma is marked by enrichment of pro-inflammatory bacterial species. Moreover, protective genera such as Dolosigranulum and Corynebacterium in the upper airways are associated with lower risk of asthma and reduced respiratory infections. Elucidating these microbiome-mediated mechanisms holds promise for the development of targeted microbiota-based strategies for asthma prevention and phenotype-specific therapeutic interventions. The present review unpacks these localized microbial patterns and their mechanistic implications for asthma development, severity, and endotypic variation. Finally, unraveling the microbiome–asthma axis from airway and gut microbial communities also has implications for new ways of thinking personalized medicine in the future.
Introduction
Asthma is a chronic, heterogeneous respiratory disease that affects over 300 million individuals globally, with increasing prevalence in both developed and developing countries (Porsbjerg et al., 2023). Clinically, asthma manifests with episodes of wheezing, breathlessness, chest tightness, and coughing, which are typically associated with variable airflow obstruction and bronchial hyperresponsiveness (Dasgupta et al., 2023). However, the disease is far from uniform—distinct clinical phenotypes and underlying molecular endotypes have been recognized, including allergic (Th2-high), nonallergic (Th2-low), eosinophilic, neutrophilic, and obesity-related asthma. This heterogeneity poses significant challenges for effective diagnosis, stratification, and management of the disease.
In recent years, there has been growing recognition of the role of the human microbiome in shaping immune responses relevant to asthma pathogenesis (Kozik and Huang, 2019; Stokholm et al., 2018). The concept of the gut–lung axis—a bidirectional communication network between the gastrointestinal and respiratory systems—has gained prominence, emphasizing how gut microbial dysbiosis can influence pulmonary inflammation. Additionally, microbial colonization of the upper and lower airways, particularly during early life, has been implicated in modulating asthma risk, severity, and therapeutic responsiveness (Lukacs and Huang, 2020).
Metagenomics has emerged as a powerful tool to decode these complex host–microbiome interactions. Unlike traditional culture-based approaches, metagenomic sequencing enables comprehensive, culture-independent profiling of microbial communities. Two primary strategies are employed: 16S ribosomal ribonucleic acid (16S rRNA) gene sequencing, which targets conserved regions of bacterial ribosomal RNA genes to infer taxonomic composition, and shotgun whole-genome sequencing, which captures a broader picture by sequencing all microbial DNA in a sample, allowing for species-level resolution and functional pathway analysis (Durazzi et al., 2021; Yen and Johnson, 2021). These approaches have uncovered previously unrecognized microbial signatures associated with asthma onset, phenotypes, and treatment response.
The aim of this expert review is to unpack, synthesize, and highlight the microbial landscape in individuals with asthma and how metagenomic technologies are reshaping our understanding of asthma. The latest findings from gut and airway microbiome studies are discussed herein, including the relevance of microbial metabolites and environmental modulators, and the potential of microbiome-based asthma signatures. Precisely, this analysis aims to provide an integrative overview of the current landscape and future directions of metagenomics in asthma research and precision medicine.
Inflammatory Endotypes of Asthma: Eosinophilic, Neutrophilic, Mixed, and Paucigranulocytic
Asthma is a heterogeneous disease, and inflammatory profiling of airway cells has enabled the classification of four major endotypes based on sputum cytology: eosinophilic, neutrophilic, mixed granulocytic, and paucigranulocytic asthma (Feng et al., 2023). Figure 1 demonstrates the endotypes of asthma and their immunological characteristics. Eosinophilic asthma is characterized by elevated eosinophils and is typically associated with Th2-high inflammation, allergic sensitization, and favorable response to corticosteroids or biologics (Hussain and Liu, 2024). Neutrophilic asthma, in contrast, shows increased airway neutrophils, often driven by Th1/Th17 pathways, and is commonly linked to severe, steroid-resistant disease and exposure to pollutants or infections (Ray and Kolls, 2017). Mixed granulocytic asthma involves elevation of both eosinophils and neutrophils, reflecting a more complex and often severe inflammatory process, and is associated with poor treatment outcomes. Paucigranulocytic asthma, defined by low levels of both eosinophils and neutrophils, may represent a noninflammatory phenotype or involve structural airway changes with limited overt inflammation (Pignatti et al., 2022).
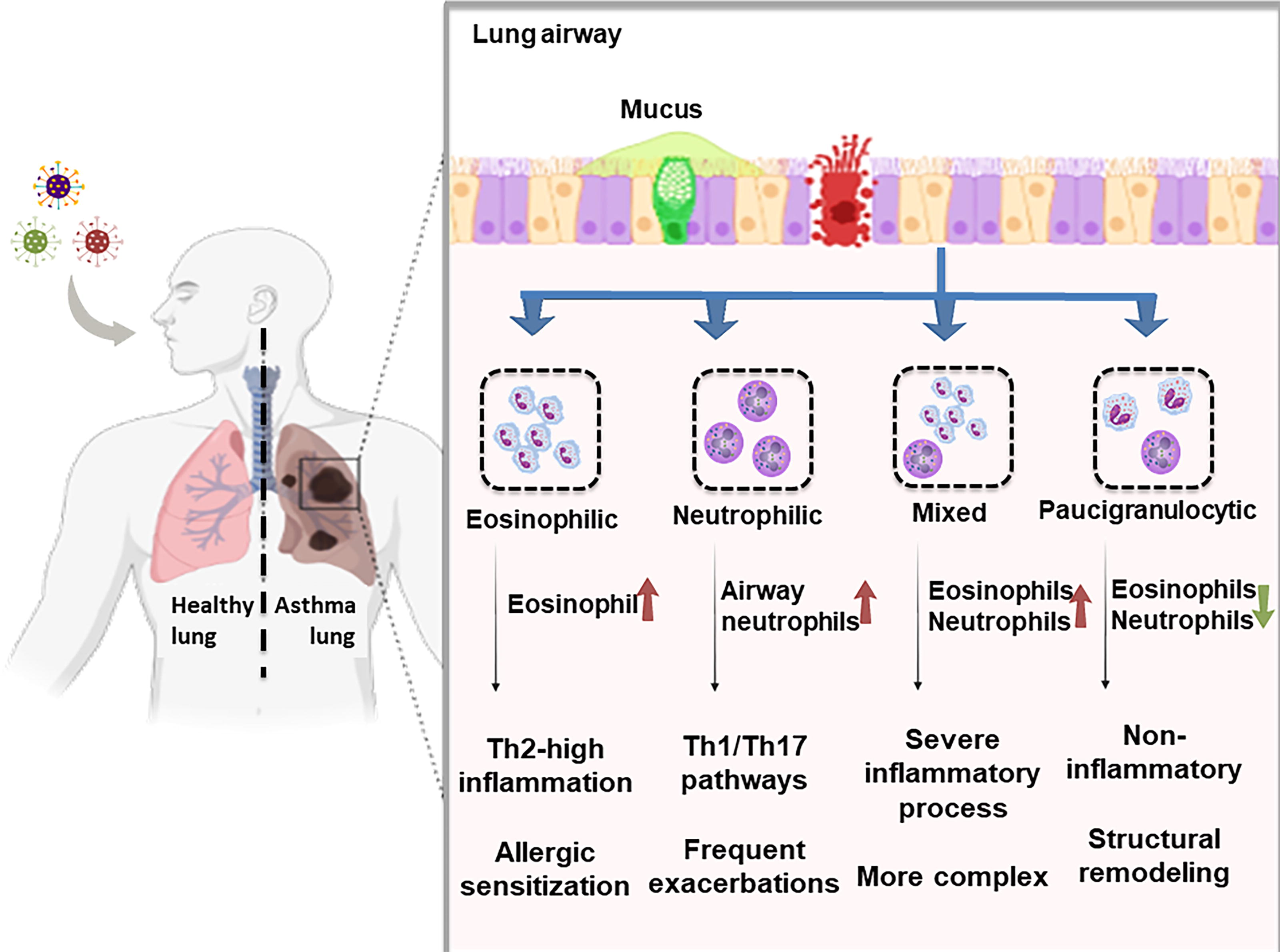
Inflammatory endotypes of asthma and their immunological characteristics. This schematic illustrates the four primary inflammatory endotypes of asthma—eosinophilic, neutrophilic, mixed granulocytic, and paucigranulocytic—based on sputum cytology and dominant immune cell infiltration. Eosinophilic asthma is typically Th2-driven and responds well to corticosteroids, whereas neutrophilic asthma involves Th1/Th17 pathways, heightened airway inflammation, and poor steroid responsiveness. Mixed granulocytic asthma reflects simultaneous eosinophil and neutrophil elevation, often associated with severe and refractory disease. Paucigranulocytic asthma shows minimal granulocyte infiltration and may reflect structural airway changes or noninflammatory mechanisms.
These endotypes are increasingly linked to distinct airway microbiome profiles, reinforcing the potential of metagenomic approaches to uncover mechanistic differences and guide precision treatment strategies. Table 1 indicates the notable microbiome composition in different asthma subtypes and their immune response.
Microbial Community Composition in Different Asthma Endotypes
IL, interleukin.
Factors Shaping Distinct Microbiota Signatures in Asthma
The human microbiome, composed of trillions of microorganisms residing in the gut, airways, and on epithelial surfaces, is fundamental to immune system development and homeostasis (Ogunrinola et al., 2020). In asthma, an immune-mediated disease marked by heterogeneous inflammatory responses, the composition and function of the microbiome play critical roles in shaping disease susceptibility and phenotype expression. A schematic diagram on the factors that influence the microbiota of asthma is shown in Figure 2.
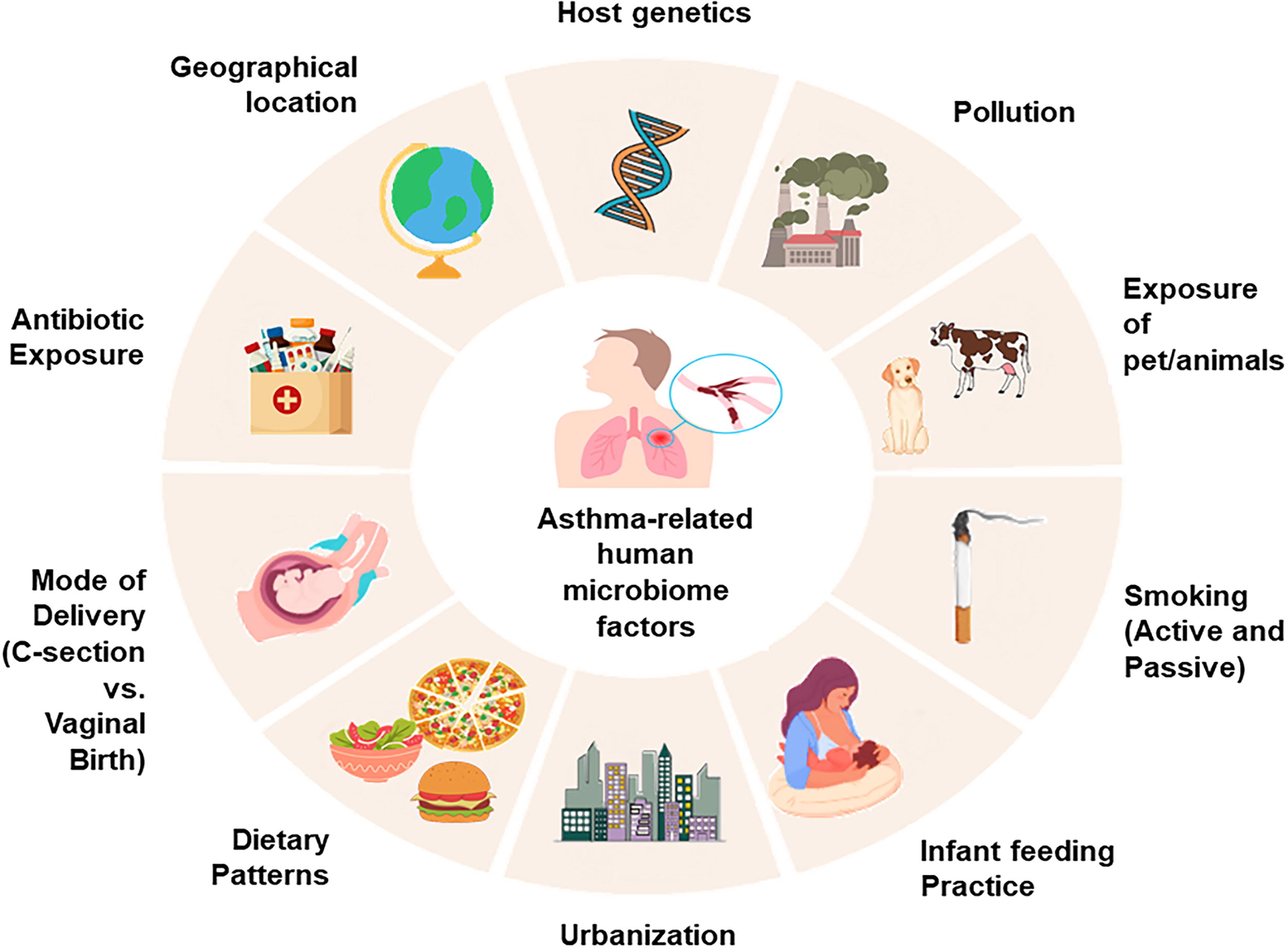
Multifactorial influences on the human microbiome relevant to asthma pathogenesis. Environmental, lifestyle, and host-related factors collectively shape the composition and function of the human microbiome, impacting immune development and asthma susceptibility. Elements such as diet, pollution, sunlight exposure, temperature, smoking, cosmetic use, pet contact, and geographic location modulate microbial diversity and community stability. Host-specific factors, including genetics and early-life exposures, further influence microbiome–immune system interactions, particularly along the gut–lung axis.
During early life, both gut and respiratory tract microbiota influence the maturation of regulatory immune cells, such as Tregs, and calibrate cytokine responses to foster immune tolerance. Disruption of this equilibrium—termed “dysbiosis”—has been increasingly associated with asthma onset, particularly when occurring during critical developmental windows. Microbial composition is not a static feature but is dynamically influenced by a network of environmental, lifestyle, and host-specific factors. Key contributors include diet (especially fiber and fat intake), antibiotic use, delivery mode, and infant feeding practices—all of which shape the early microbiome and its immune training capacity. Additional factors—such as air pollution, smoking, geographic location, climate, pet or farm animal exposure, urbanization, and host genetics—further modulate microbial diversity and function across the lifespan. These influences converge to affect the gut–lung axis, a key conduit of immunological signaling between distal mucosal sites. Importantly, studies have also reported an association between reduced microbial richness and increased asthma prevalence, supporting the hygiene hypothesis (Loverdos et al., 2019; Zhang et al., 2024).
The gut–lung axis operates through several interconnected mechanisms that enable gut-resident microbes to modulate immune responses at distal mucosal sites, including the lungs. Microbial metabolites such as short-chain fatty acids (SCFAs)—notably acetate, propionate, and butyrate—produced by gut commensals, enter systemic circulation and influence pulmonary immunity by enhancing epithelial barrier integrity, promoting the differentiation of regulatory T cells (Tregs), and suppressing proinflammatory cytokine production (Sasaki et al., 2024; Silva et al., 2020; Zhang et al., 2023). Concurrently, microbial-associated molecular patterns interact with host pattern recognition receptors, particularly Toll-like receptors (TLRs), initiating signaling cascades that modulate immune tone and foster mucosal tolerance (Rhee SH, 2011). Dysbiosis in the gut can impair these regulatory pathways, skewing immune responses toward Th2- and Th17-dominated inflammation—hallmarks of asthma (Barcik et al., 2020; Toor et al., 2019). Additionally, immune cells primed within gut-associated lymphoid tissues may traffic to the lungs, further contributing to local immune modulation. These cross-compartmental interactions highlight the systemic influence of gut microbiota on respiratory health and underscore their mechanistic role in asthma pathogenesis.
Environmental influences on the human microbiome and implications for asthma susceptibility
The environment plays a pivotal role in shaping the composition and function of the human microbiome, beginning from early life and continuing throughout the lifespan. The human microbiome is essential for maintaining host physiology, contributing to key processes such as nutrient metabolism, immune system education, and defense against pathogens. However, these microbial functions are not static; they are profoundly influenced by environmental exposures at both macro and micro levels (Ahn and Hayes, 2021). Macroenvironmental factors—such as urbanization, air pollution, chemical exposures, and socioeconomic disparities—can alter microbial communities, potentially inducing dysbiosis and increasing susceptibility to chronic inflammatory diseases, including asthma. Concurrently, microenvironmental factors at the individual level—such as diet, smoking, alcohol use, hygiene practices, and antibiotic exposure—shape microbial diversity and functionality. For example, Western-style diets low in fiber and high in fat have been linked to decreased production of SCFAs, impairing epithelial barrier integrity and immune regulation. In contrast, early-life exposures to pets or farm environments may facilitate colonization by beneficial microbes and lower asthma risk. These complex, bidirectional interactions between the environment, microbiome, and host highlight the importance of integrated, longitudinal research to identify modifiable factors and inform preventive strategies for asthma and related noncommunicable diseases. Table 2 summarizes key environmental factors that influence the human microbiome and outlines their relevance to asthma pathogenesis.
Environmental Factors Influencing the Human Microbiome and Their Relevance to Asthma Pathogenesis
RSV, respiratory syncytial virus; SCFA, short-chain fatty acid.
Dysbiosis in asthma
Asthmatic individuals frequently exhibit altered microbial profiles characterized by reduced alpha diversity and an imbalance between beneficial and proinflammatory taxa. In the airways, dysbiosis is commonly marked by an overrepresentation of potentially pathogenic genera such as Haemophilus, Moraxella, Streptococcus, Neisseria, and Klebsiella. This microbial enrichment is associated with neutrophilic inflammation and resistance to corticosteroid therapy (Green et al., 2014), particularly in non-eosinophilic asthma phenotypes. Concurrently, beneficial taxa such as Lactobacillus and members of Mogobacteriaceae are depleted, potentially impairing mucosal immunity and barrier integrity.
Gut microbiota in asthma similarly exhibit imbalances, including increased levels of Streptococcus and Bacteroides, alongside a marked reduction in anti-inflammatory and immunoregulatory genera such as Bifidobacterium, Faecalibacterium, and Akkermansia. These changes are correlated with greater epithelial permeability, allergic sensitization, and frequency of asthma exacerbations, especially when dysbiosis occurs during early childhood—a critical window for immune development. Fungal dysbiosis also contributes to asthma pathophysiology. In the lungs, the presence of Malassezia and Alternaria has been associated with enhanced allergic airway inflammation. In the gut, increased abundance of fungi such as Candida and Rhodotorula has been linked to a higher risk of asthma, particularly in pediatric populations (Kanj and Skalski, 2024). Experimental models confirm that certain fungal species can exacerbate lung inflammation by modulating immune pathways. Viral exposures further complicate the microbial landscape in asthma. Respiratory viruses including rhinovirus, respiratory syncytial virus (RSV), and influenza are strongly implicated in both asthma onset and exacerbations, often interacting with bacterial and fungal communities to amplify airway inflammation (Barcik et al., 2020; Ortega et al., 2021). Table 3 highlights the microorganisms in the lungs and gut linked to asthma-associated dysbiosis.
Microorganisms in the Lungs and Gut Linked to Asthma-Associated Dysbiosis
Impact of early-life microbial exposures
The first 1000 days of life represent a critical window in which microbial exposures have long-term effects on immune development and asthma risk. Vaginal birth, breastfeeding, pet exposure, and absence of antibiotics during infancy favor the establishment of a diverse and protective microbiota. In contrast, caesarean delivery, early antibiotic exposure, and sanitized living environments hinder microbial maturation and increase asthma susceptibility by impairing immune tolerance and gut–lung signaling (Kahhaleh et al., 2024).
Gut microbial profiles and later asthma development
Longitudinal birth cohort studies (e.g., Canadian Healthy Infant Longitudinal Development [CHILD], Kind, Ouders en gezondheid: Aandacht voor Leefstijl en Aanleg {Translation: Child, Parent and Health: Lifestyle and Genetic Constitution} [KOALA]) have linked early-life gut microbial composition to later asthma development (Penders et al., 2007; Subbarao et al., 2015). Infants with reduced levels of Lachnospira, Veillonella, Faecalibacterium, and Rothia within the first 3 months of life show a greater likelihood of developing asthma during childhood (Arrieta et al., 2015). Functional metagenomic analyses reveal that these taxa contribute to the production of SCFAs and modulate immune pathways related to epithelial repair and inflammation control. These findings underscore the potential of early microbial profiling for asthma prediction and preventive intervention.
Metagenomic Approaches in Asthma Research
As mentioned earlier, metagenomics has revolutionized microbiome research by enabling comprehensive, culture-independent profiling of microbial communities in human health and disease. In asthma research, metagenomic techniques have provided unprecedented insights into the composition, diversity, and functional potential of airway and gut microbiota. This section outlines the typical laboratory and computational workflows involved in metagenomic studies, key tools and databases, and the advantages of these approaches over traditional methods.
Laboratory workflow: From sample to sequencing
Characterizing the human microbiome in asthma requires carefully chosen sampling methods that balance invasiveness, anatomical relevance, and microbial load. For gut microbiome analysis, stool samples are the standard, offering noninvasive collection and rich microbial content suitable for both 16S rRNA and shotgun metagenomic sequencing. However, fecal content does not directly reflect local interactions between, for example, intestinal epithelial cells and microbiota (Didriksen et al., 2024), which may be best studied with direct sampling that is more feasible in animal models. In contrast, airway microbiome sampling presents unique challenges due to the low biomass and potential for contamination. Common upper airway samples include nasal swabs, nasopharyngeal aspirates, and oropharyngeal swabs, which are minimally invasive and often used in pediatric studies. For lower airway profiling, induced sputum and bronchoalveolar lavage fluid (BALF) provide deeper insights but vary in accessibility and invasiveness, with BALF being more precise but typically limited to hospital settings. Exhaled breath condensate (EBC) and saliva are emerging as complementary noninvasive matrices for microbiome and metabolite profiling of complex lung diseases (Choudhury et al., 2021; Armstrong et al, 2021). It is also important to note that microbial compositions differ between samples of the upper and lower respiratory tract (Bassis et al., 2015; Durack et al., 2018; Huang, 2025). Standardization of collection protocols, storage conditions, and DNA extraction methods is crucial to ensure reproducibility and comparability across asthma microbiome studies.
Comparative scope: 16S rRNA versus shotgun metagenomics
Metagenomic studies in asthma have primarily employed two approaches: 16S rRNA gene sequencing and shotgun metagenomics. The 16S rRNA method targets a conserved bacterial gene, allowing for taxonomic identification—usually at the genus level—with relatively low cost and computational demand. While useful for assessing microbial diversity and broad community shifts, it offers limited resolution and excludes nonbacterial domains such as viruses and fungi. In contrast, shotgun metagenomics sequences all DNA in a sample, enabling species- and strain-level identification across all microbial kingdoms, along with direct functional profiling of genes and pathways. Although more expensive and data-intensive, shotgun metagenomics provides a comprehensive view of both taxonomic composition and metabolic potential—crucial for understanding host–microbe interactions, immune modulation, and microbial contributions to asthma heterogeneity. As asthma research increasingly prioritizes mechanistic insights and precision medicine, shotgun metagenomics is emerging as a powerful tool to uncover novel biomarkers and therapeutic targets. While 16S rRNA sequencing remains cost-effective and widely used, shotgun metagenomics is increasingly preferred in asthma research due to its ability to identify nonbacterial taxa and functional gene pathways relevant to host–microbiome interactions. Table 4 presents a detailed comparison of these two sequencing approaches, highlighting their respective advantages, limitations, and applications in the context of microbiome research.
Comparative Overview of 16S rRNA Sequencing and Shotgun Metagenomics: Advantages, Limitations, and Applications in Microbiome Research
16S rRNA, 16S ribosomal ribonucleic acid.
Key bioinformatics tools and pipelines
Several bioinformatics tools are widely used in microbiome research to process, analyze, and interpret sequencing data derived from 16S rRNA and shotgun metagenomics.
Apart from these bioinformatics-based tools, reference databases are also used in microbiome research for taxonomic classification and functional annotation. SILVA and Greengenes provide well-curated 16S rRNA gene databases, while GTDB offers standardized bacterial and archaeal taxonomy based on whole-genome data. IMG/M supports comparative functional genomics with access to curated microbial genomes. Additionally, National Center for Biotechnology Information (NCBI)-RefSeq and MGnify are frequently used for sequence alignment and annotation, enhancing the accuracy of metagenomic analyses (Balvočiūtė and Huson, 2017).
Strengths of metagenomics
Compared with traditional culture-based techniques, which can only recover a small fraction of microbial taxa, metagenomics enables detection of unculturable and low-abundance organisms (Chen et al., 2022). Additionally, shotgun metagenomics offers significant advantages over 16S-only methods by enabling:
By integrating both taxonomic and functional insights, metagenomics represents a transformative approach in asthma research, offering a more holistic understanding of host–microbiome interactions and their role in disease phenotypes and progression.
Key Findings from Metagenomic Studies in Asthma
Metagenomic research has significantly advanced our understanding of asthma pathogenesis, particularly by elucidating the relationships between microbial communities, immune system development, and asthma phenotypes. This section highlights major insights gained from both taxonomic and functional metagenomic analyses of the gut and airway microbiome.
Gut microbiota and asthma onset
Longitudinal birth cohort studies have been pivotal in linking early-life gut microbial composition to subsequent asthma risk. Two major studies in this area include:
These studies have emphasized that protective taxa, including Bifidobacterium and Faecalibacterium, promote immune tolerance via the production of anti-inflammatory metabolites such as SCFAs. In contrast, early dysbiosis—characterized by low microbial richness and loss of beneficial taxa—can impair the development of regulatory T cells and favor a Th2-skewed immune response, predisposing to allergic asthma.
The nasopharyngeal and respiratory microbiome in asthma phenotypes
Unlike the gut, the lower airway harbors less diverse but clinically significant microbiota that modulate local immune responses and contribute to asthma heterogeneity. Distinct microbial patterns have been observed across asthma phenotypes. In eosinophilic asthma, the airway microbiome often resembles that of healthy individuals and is typically enriched with Prevotella species, which may exert anti-inflammatory effects (Campbell et al., 2023). In contrast, neutrophilic asthma is characterized by increased abundance of potentially pathogenic bacteria such as Haemophilus influenzae, Moraxella catarrhalis, and Streptococcus pneumoniae. These taxa can trigger TLR signaling pathways, leading to the production of proinflammatory cytokines such as interleukin-8 and promoting neutrophilic infiltration (Versi et al., 2023). Notably, the presence of Dolosigranulum and Corynebacterium in the upper airway is associated with reduced susceptibility to respiratory infections and asthma development, with Corynebacterium also linked to lower eosinophilic inflammation in adults (Kozik and Huang, 2019). Collectively, these observations underscore the compartment-specific influence of airway microbiota on immune regulation and asthma pathogenesis, offering potential targets for phenotype-specific interventions.
Limitations and Challenges in Metagenomic Asthma Research
Despite its transformative potential, metagenomic research in asthma faces several critical limitations that constrain its interpretability, reproducibility, and clinical translation. One of the most pressing technical challenges lies in the analysis of low-biomass respiratory samples—such as sputum, nasal swabs, or BALF—which are highly susceptible to contamination from reagents and the laboratory environment. This contamination can lead to misleading microbial profiles, especially when true residents of the airways exist in low abundance (Dulanto Chiang and Dekker, 2020). Additionally, limited sequencing depth may prevent detection of rare, yet functionally significant, microbial taxa or genes. In shotgun metagenomics, where both taxonomic resolution and functional profiling are desired, high sequencing coverage becomes essential, further increasing costs and technical complexity.
Biological and methodological variabilities further complicate metagenomic investigations in asthma. The human microbiome is inherently dynamic, shaped by diet, medication use, infections, hormonal cycles, geography, and host-specific factors such as age and comorbidities. These sources of intra- and inter-individual variabilities make it difficult to identify consistent microbial patterns across different populations or studies (Forry et al., 2024). Compounding this issue is the widespread use of cross-sectional study designs, which offer limited insight into causality. Many studies also suffer from small sample sizes and lack representation from diverse ethnic, geographic, and socioeconomic backgrounds, reducing the generalizability of findings. Technical inconsistencies in sample collection, storage, and processing introduce additional batch effects that can obscure true biological signals.
A critical translational gap also remains between metagenomic associations and mechanistic understanding of asthma pathophysiology. While metagenomic profiling can identify microbial shifts linked to specific asthma endotypes or severity, functional validation is often lacking. Experimental systems—such as gnotobiotic (germ-free or humanized) animal models and human-derived organoids—are urgently needed to test the causal roles of candidate microbes, metabolites, and microbial genes (Gulliver et al., 2022). Reproducibility is further limited by variability in computational workflows, including differences in bioinformatics pipelines (e.g., QIIME2, MetaPhlAn, HUMAnN), reference databases (e.g., SILVA, Greengenes), and statistical methodologies. Standardized, transparent, and interoperable protocols are essential for ensuring consistency, facilitating data sharing, and accelerating the clinical translation of microbiome-based insights in asthma research.
Future Directions
As asthma research advances and edges toward precision/personalized medicine, metagenomics will play a central role in multi-dimensional frameworks that integrate diverse “omics” data and technological innovations. A key future direction is the convergence of metagenomics with metabolomics, transcriptomics, proteomics, epigenomics, and immunomics to capture the full complexity of host–microbe–environment interactions. These integrative approaches can provide mechanistic insights into asthma heterogeneity by linking microbial community structure with host gene expression, immune signaling, and metabolite production. Artificial intelligence and machine learning models are poised to revolutionize biological databases by identifying predictive signatures of disease risk, overlapping phenotypic features, and individualized treatment responses (Dasgupta, 2024). Combining metagenomic features with clinical, environmental, and molecular data can enable robust, personalized interventions and facilitate early risk prediction.
Looking ahead, longitudinal and multiethnic cohort studies are essential to capture microbiome dynamics across developmental windows and diverse populations. Harmonized sampling and metadata protocols will enhance data comparability and enable large-scale meta-analyses through global consortia. Parallel advances in synthetic biology—including CRISPR-based gene editing, phage therapy, and designer probiotics—offer exciting opportunities to rationally manipulate the microbiome for therapeutic benefit. However, these innovations must be evaluated through rigorous safety and efficacy studies, especially in pediatric populations. Ethical and regulatory frameworks must also evolve to address concerns around microbiome data privacy, consent, product classification, and equitable access to emerging microbiome-based diagnostics and therapeutics. Responsible innovation, underpinned by global policy alignment and public engagement, will be key to translating microbiome science into clinical solutions for asthma.
Conclusions
Advances in metagenomics have transformed our understanding of asthma, revealing that microbial communities—particularly in the gut and airways—play a critical role in disease onset, progression, and phenotypic variability. These insights underscore the biological complexity and heterogeneity of asthma, moving beyond a one-size-fits-all model toward a more nuanced, endotype-driven framework of disease.
By leveraging high-resolution sequencing technologies and functional profiling tools, research thus far has identified the key microbial taxa and metabolites—such as Faecalibacterium, Bifidobacterium, and SCFA—that modulate immune tolerance and inflammation. Additionally, metagenomic comparisons across asthma phenotypes have revealed distinct microbial signatures associated with eosinophilic versus neutrophilic inflammation and treatment responsiveness. These findings not only deepen our mechanistic understanding but also open new avenues for noninvasive diagnostics, risk stratification, and microbiome-targeted interventions.
On the contrary, despite its promise, metagenomics in asthma research is still evolving and emerging. Technical challenges, limited population diversity in the available datasets, and translational gaps remain as formidable barriers to achieving real-life clinical applications. Finally, unraveling the microbiome–asthma axis from airway and gut microbial communities also has implications for new ways of thinking personalized medicine in the future. Moving forward, collaborative, interdisciplinary efforts are essential—uniting microbiologists, immunologists, clinicians, computational scientists, ethicists, and social theorists—to translate microbiome insights into personalized therapeutics and preventative strategies. Ultimately, metagenomics holds immense potential to redefine asthma care by enabling early prediction, mechanistic stratification, and precision/personalized treatment based on an individual’s microbial landscape.
Footnotes
Author’s Contributions
S.D.: Conceptualization, formal analysis, investigation, visualization, and writing—review and editing.
Author Disclosure Statement
The author declares she has no conflicting financial interests.
Funding Information
No funding was received for the present study.