
Abstract
Endometrial carcinoma (EC) presents a growing global health challenge, characterized by significant molecular heterogeneity and the overexpression of the mitotic motor protein Kinesin Family Member 11 (KIF11). To address the critical need for targeted therapeutics, this study employed an integrated computational framework combining multiomics expression analysis with structural bioinformatics to identify novel natural product-based inhibitors. Validation using TCGA and CPTAC datasets confirmed significant upregulation of KIF11 in EC at both transcriptomic and proteomic levels. A pharmacophore-based virtual screening of approximately 400,000 natural compounds was conducted against the KIF11 active site (PDB ID: 2X7C), followed by ADMET profiling and molecular docking. While glycodeoxycholic acid (GDCA) exhibited the highest docking affinity (−9.5 kcal/mol), subsequent 100 ns molecular dynamics simulations revealed that glycocholic acid (GCA) possessed superior conformational stability. GCA demonstrated a stable RMSD profile (approximately 1.5 Å) and persistent hydrogen bonding with key residues LYS111 and GLU118, unlike the fluctuating profile observed in GDCA. This study identifies GCA as a robust lead candidate, underscoring the efficacy of integrating transcriptomic and proteomic data with structural biology to drive precision therapeutics for EC.
Introduction
Endometrial carcinoma (EC) stands as a significant global health challenge, representing the third most common gynecological cancer among women aged 50–70 worldwide, according to GLOBOCAN 2022 data (Ferlay et al., 2021; Sung et al., 2021). While prevalent globally, it disproportionately affects postmenopausal women in developed countries. With a staggering 662,301 reported incidences and 348,874 deaths worldwide, the substantial burden of EC underscores an urgent need for advanced understanding and more effective treatment strategies (Ferlay et al., 2021; Sung et al., 2021).
The inherent diversity and heterogeneity of EC cells, characterized by multiple subtypes, chromosomal abnormalities, and altered molecular pathways, present considerable challenges in both treatment efficacy and prognostic accuracy (Hou et al., 2020). Current standard treatments for EC encompass surgery, radiation, chemotherapy, and hormone therapy (Crosbie et al., 2022). However, persistent issues such as financial constraints, the rigor of clinical trials, high recurrence rates, and debilitating side effects highlight a critical need for more precise, targeted therapeutic approaches (Bernards et al., 2020).
In response to this unmet clinical need, the ongoing development of targeted therapies is revolutionizing drug discovery. Pharmacophore-based virtual screening, in particular, has emerged as a pivotal tool for identifying potent inhibitors of specific therapeutic targets (Zhou et al., 2019). This technique offers compelling advantages through its simplicity, versatility, and efficiency, primarily by focusing on the essential three-dimensional arrangement of molecular features required for biological activity (Zhou et al., 2019). This targeted approach facilitates rapid screening of vast chemical libraries, enabling scaffold hopping and the discovery of novel compounds with desired biological effects (Zhou et al., 2019). Complementing this, natural products represent a rich and historically successful source of therapeutic candidates. Derived from plants, fungi, and bacteria, natural products frequently exhibit unique mechanisms of action, superior selectivity, and reduced toxicity compared with many synthetic drugs (Romano and Tatonetti, 2019; Bailon-Moscoso et al., 2017). Given the increasing global interest in sustainable and innovative anticancer therapies, bioactive natural compounds offer a highly promising avenue for developing targeted inhibitors to improve EC management (Romano and Tatonetti, 2019; Bailon-Moscoso et al., 2017).
The present study is focused on Kinesin Family Member 11 (KIF11), also known as Eg5, identifying its 3D structure (Protein Data Bank [PDB] ID 2X7C) as a relevant therapeutic protein. KIF11 is a crucial plus-end-directed kinesin motor protein indispensable for the formation of the bipolar spindle during metaphase, a vital step in cell division (Wang et al., 2025). Its significance as a therapeutic target stems from its frequent overexpression in numerous proliferative tissues, including various cancers, while being minimally expressed in nonproliferative normal cells (Wang et al., 2020). This differential expression makes KIF11 an attractive target, as its inhibition can selectively disrupt cancer cell proliferation without severely impacting healthy cells (Guo et al., 2022). Studies have shown KIF11 to be involved in the progression of several cancer types, including prostate, colorectal, gastric, breast, and meningioma cancers, where its upregulation often correlates with disease progression (Gao et al., 2024). Inhibitors of KIF11 can block cell cycle progression and induce cell death, offering a promising strategy for anticancer therapy with potentially fewer neuropathic side effects compared with conventional antimitotic drugs (Gao et al., 2024).
In this study, pharmacophore-based virtual screening of a comprehensive natural products library was employed to identify potential KIF11 inhibitors. This was followed by a rigorous molecular docking and molecular dynamics (MD) simulation to thoroughly characterize the binding affinities and structural stability between KIF11 and the most promising inhibitors. Notably, our investigation identified a bile acid exhibiting particularly promising inhibitory interactions and sustained conformational stability within the KIF11 active site. Ultimately, this work aims to elucidate the intricate molecular mechanisms driving EC development and progression, thereby contributing to the identification of potent natural product-based inhibitors for EC management.
Materials and Methods
KIF11 expression analysis
To evaluate whether the KIF11 gene and KIF11 protein are overexpressed, the expression of KIF11 in EC was analyzed at both the messenger RNA (mRNA) and protein levels using publicly available TCGA and CPTAC datasets (https://proteomics.cancer.gov/programs/cptac; accessed on March 5, 2025; Edwards et al., 2015). A bioinformatics approach was used, employing the GEPIA2 web server (http://gepia2.cancer-pku.cn/; accessed on March 26, 2025) to compare KIF11 mRNA expression in tumor versus normal tissues via a box plot with a log2 (TPM + 1) scale, and a one-way analysis of variance to determine statistical significance (Tang et al., 2017). Concurrently, the UALCAN portal (https://ualcan.path.uab.edu/analysis-prot.html; accessed on March 27, 2025) was utilized for protein expression analysis, visualizing KIF11 protein levels in a similar box-plot format (Chandrashekar et al., 2022).
Protein retrieval and preparation
Depending on the dependency score and prognostic value of hub genes, the protein corresponding to KIF11 was selected as the target protein for further identification of a potent inhibitor. The X-ray crystal structure of KIF11 was retrieved from PDB (https://www.rcsb.org/; accessed on March 29, 2025) with PDB ID 2X7C. The 3D complex structure includes adenosine diphosphate (ADP), a magnesium ion, and S-enastron in its catalytic and allosteric sites, respectively. Nonprotein elements were removed to prevent interference during the docking process. Using AutoDock Vina’s MGL Tool (version 1.5.7), polar hydrogens were added, Kollman charges were assigned, and crystallographic waters were removed (Eberhardt et al., 2021). The resulting structure, saved in pdbqt format, was further refined by predefining the ligand binding site with BIOVIA Discovery Studio Visualizer to optimize docking studies.
Pharmacophore-based screening and library preparation
Approximately 400,000 natural compounds from the COCONUT database (https://coconut.naturalproducts.net/; accessed on April 6, 2025) were screened using a structure-based pharmacophore model of the KIF11–ADP complex (PDB ID: 2X7C) generated with PharmIT (https://pharmit.csb.pitt.edu/search.html; accessed on April 6, 2025) (Chandrasekhar et al., 2025; Sunseri and Koes, 2016). For the pharmacophore model, one aromatic ring and six hydrogen bond acceptors at a particular orientation and XYZ co-ordinates were selected and compounds with similar structural characteristics based on the model were identified. The compounds passed through pharmacophore-based screening were further filtered using Lipinski’s rule within the module, “Hit Screening” of PharmIT, followed by single conformer selection and energy minimization using Vina score (Eberhardt et al., 2021). The selected compounds, provided in SDF format, underwent 3D protonation and were converted to PDBQT format using OpenBabel. The docking simulations were executed using AutoDock Vina via Linux command-line protocols to evaluate their binding affinities.
Prediction of drug-likeness
ADMETlab 3.0 rapidly and accurately predicts a compound’s ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties, along with other crucial pharmaceutical characteristics such as mutagenicity, carcinogenicity, and physicochemical properties. ADMETlab 3.0 (https://admetlab3.scbdd.com/server/screening; accessed on April 8, 2025) evaluated screened compounds based on established pharmaceutical guidelines, including Lipinski’s Rule of Five for drug-likeness, Veber’s rules, and other key properties related to ADMET, which sets specific criteria for drug-like molecules (Fu et al., 2024).
Molecular docking studies
A virtual screening approach was employed to evaluate the binding potential of the library of compounds along with ADP against the active site of the KIF11 protein. AutoDock Vina was used for this purpose, with input files prepared according to the software’s specifications. The grid box, encompassing the active-site residues, was defined using the MGLTools, with dimensions of 46 Å, 50 Å, and 94 Å along the X, Y, and Z axes, respectively. Docking was performed using default parameters for energy range (4 kcal/mol) and exhaustiveness, resulting in eight distinct poses for each ligand. The pose exhibiting the most favorable binding profile (most negative score) was extracted for further analysis. PyMOL (Version 2.4.0, Schrödinger, LLC; 2024) was used to isolate the best-docked ligand conformation. BIOVIA Discovery Studio Visualizer facilitated the visualization and analysis of key interactions between the ligand and the KIF11 active site, including conventional hydrogen bonds, pi-alkyl interactions, and various carbon–hydrogen interactions. Compounds demonstrating the most promising interactions were selected for further evaluation.
MD simulation of complexes
The interactions between the top three ligands, which were selected based on ADMET properties and docking score, were then evaluated through MD simulations conducted using Schrödinger’s Desmond software (Version 2019. D. E. Shaw Research; 2019). CHARMM-36 all-atom force field was employed to generate the protein’s topology file, while the CGenFF online service (https://cgenff.com/; accessed on May 10, 2025) was utilized to prepare the topology files for the ligands. The protein–ligand complex was solvated using a TIP3P water model, and the system was neutralized with the appropriate amount of Na+ and Cl− ions. Energy minimization was performed using the gradient descent optimization algorithm to stabilize the system. Prior to the 100 ns production phase, the system underwent 100 ps of equilibration in both NVT (constant number of particles, volume, and temperature) and NPT (constant number of particles, pressure, and temperature) ensembles. Postproduction, the trajectory was analyzed for root mean square deviation (RMSD), root mean square fluctuations (RMSF), the total number of hydrogen bonds, and binding energies between the ligands and the protein.
Results
Expression of KIF11 in EC
Analysis of mRNA expression using the GEPIA2 server, derived from TCGA data, revealed a statistically significant upregulation of KIF11 in EC tissues compared with normal controls (Fig. 1A). Correspondingly, differential protein expression analysis conducted via the UALCAN portal, utilizing CPTAC data, also demonstrated a significant upregulation of KIF11 protein levels in EC (Fig. 1B).
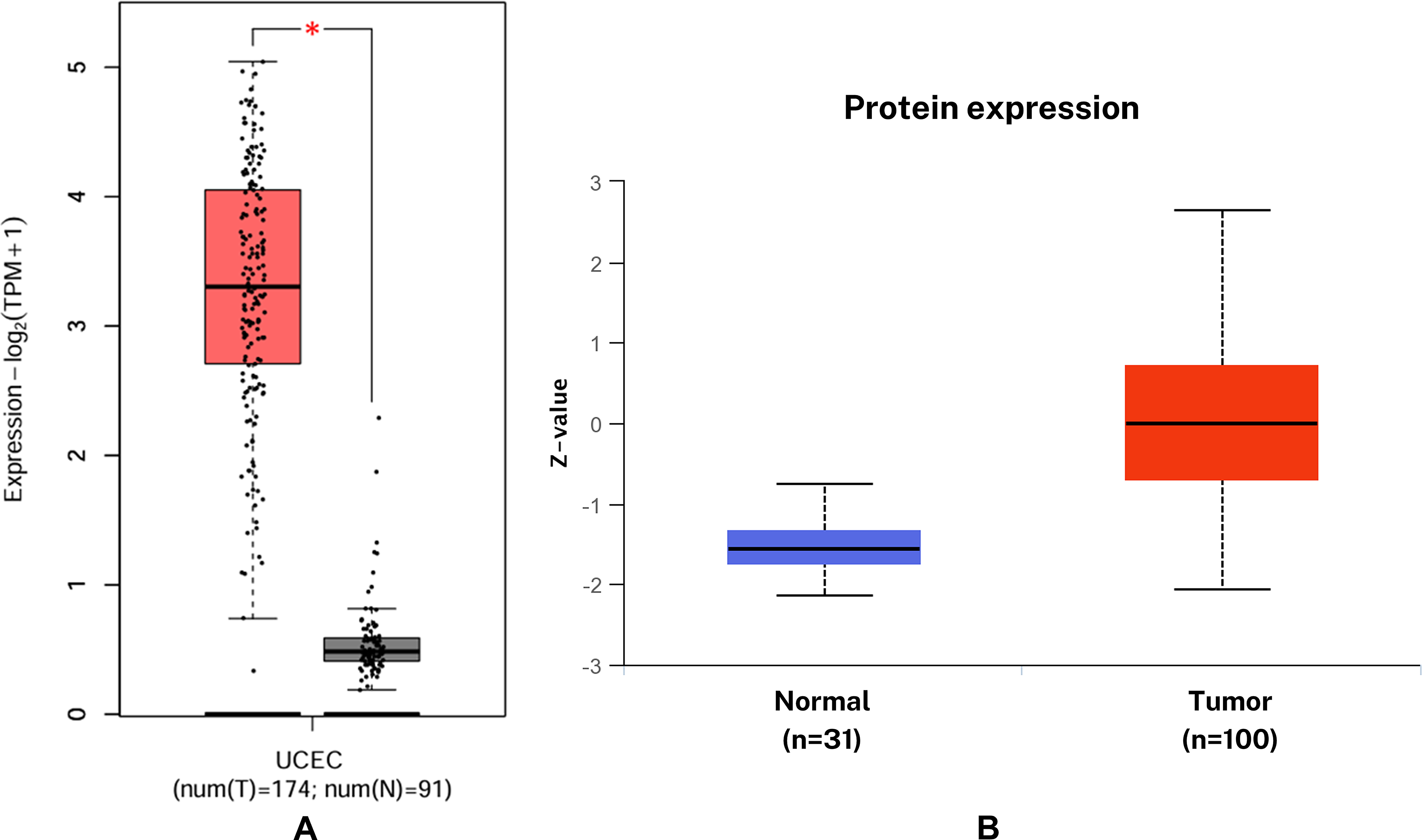
The expression of KIF11 in EC:
Screening of natural product compounds
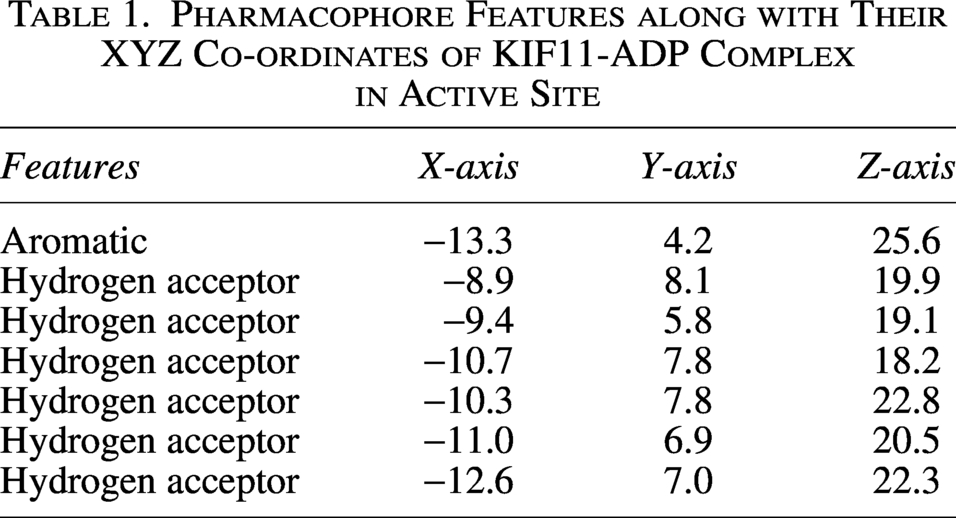
An extensive library of approximately 400,000 compounds was screened for potential interactions with the target protein KIF11. To identify promising interacting candidates, structure-based pharmacophore features (Table 1 and Fig. 2A and B) were further used as filters for screening the library, resulting in 2,516 compounds. With the various stages of rigorous assessment, based on Lipinski’s rule of five, thousands of compounds were narrowed down to 81 compounds that were exhibiting favorable drug-like characteristics. However, as the library contained more than one conformer for a single compound, additional constraints were applied to remove multiple conformers and leaving only 56 compounds.
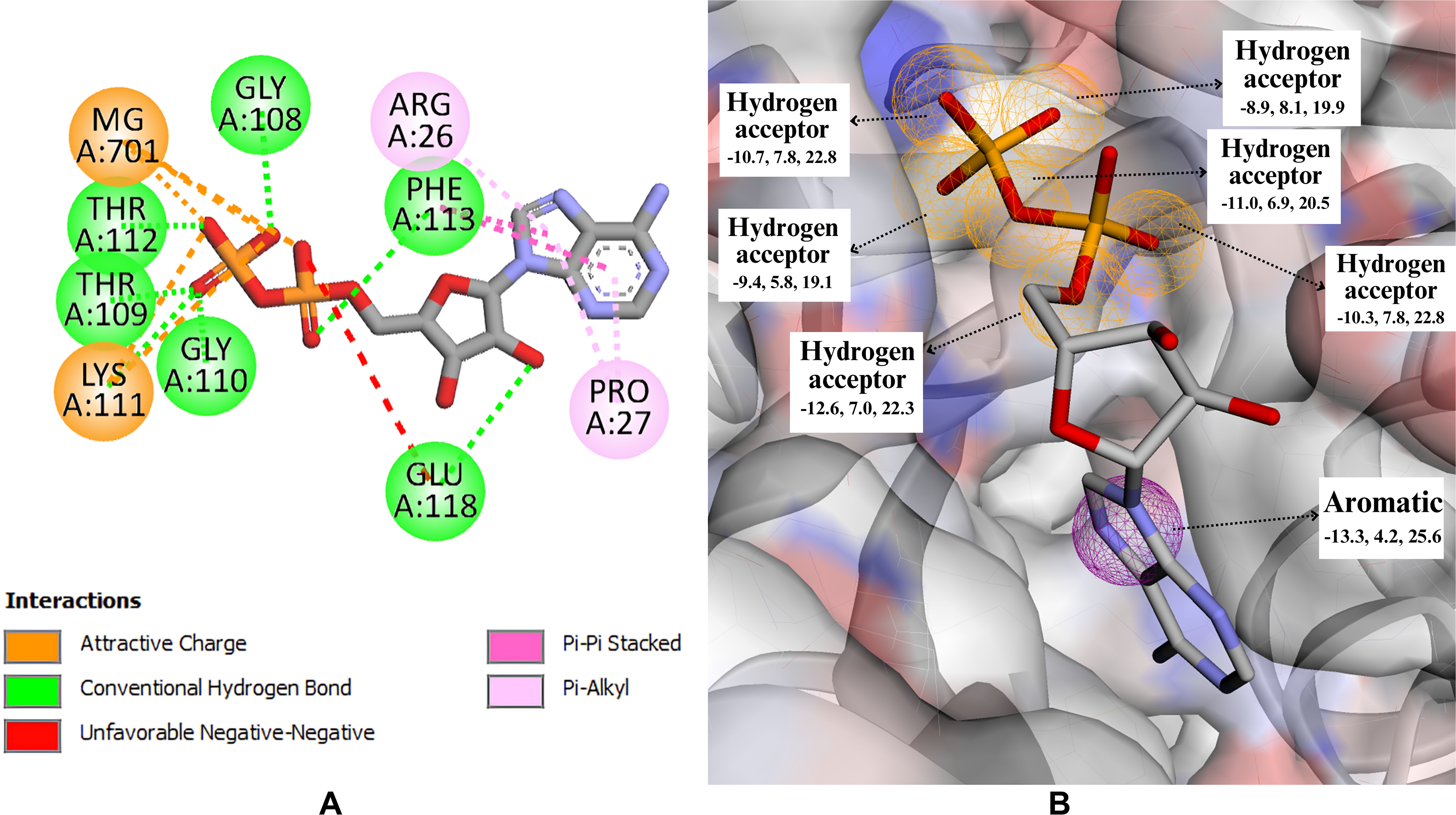
The figure depicts the molecular interactions between a native ligand ADP and KIF11.
Pharmacophore Features along with Their XYZ Co-ordinates of KIF11-ADP Complex in Active Site
Pharmacokinetic analysis
ADMETlab3.0 was used to evaluate the physicochemical properties, pharmacokinetic profiles, toxicity, and synthetic accessibility of the 56 filtered compounds. Based on the ADMET predictions, these compounds exhibit a highly desirable set of safety and pharmacological properties. Both glycocholic acid (GCA) and glycodeoxycholic acid (GDCA) show excellent safety profiles with no predicted toxicophores and a low risk of hERG (human-ether-à-go-go-related gene)-related cardiotoxicity (Table 2). Their minimal intestinal absorption and limited ability to cross the blood–brain barrier are valuable traits, suggesting potential for localized therapeutic action without systemic or central nervous system (CNS) side effects. Eleven-oxo-androsterone glucuronide (OAG) stands out with an exceptionally clean safety profile, including an extremely low hERG risk. Its high hydrophilicity and minimal absorption mean it is unlikely to cause systemic side effects, and its slow clearance rate allows for a sustained presence for targeted delivery.
Predicted ADMET Properties of GCA, GDCA, and OAG
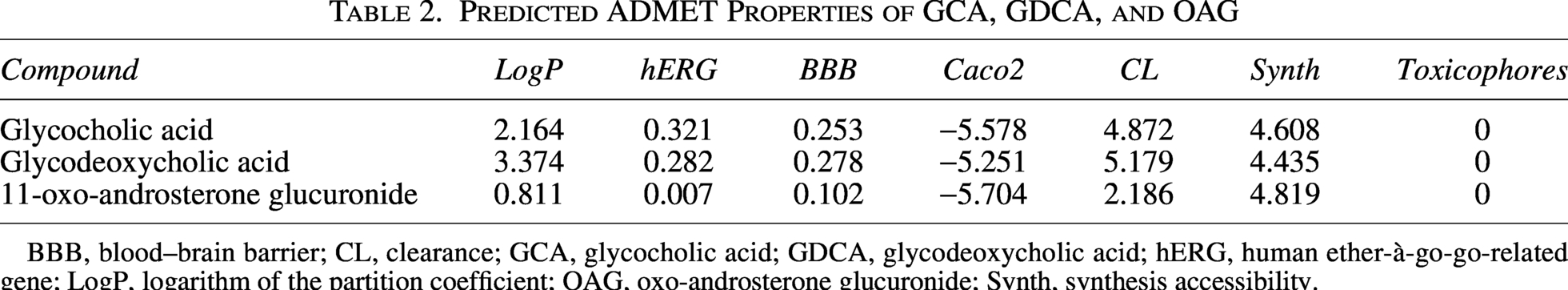
BBB, blood–brain barrier; CL, clearance; GCA, glycocholic acid; GDCA, glycodeoxycholic acid; hERG, human ether-à-go-go-related gene; LogP, logarithm of the partition coefficient; OAG, oxo-androsterone glucuronide; Synth, synthesis accessibility.
Molecular interactions and binding mode of potential compounds
Following the initial filtering to 56 compounds (Supplementary Table S1), a stringent multiparameter selection criteria were applied to identify the most promising candidates for MD simulations. Rather than relying solely on docking scores, candidates were prioritized based on a combined optimization of their AutoDock Vina docking scores, synthetic accessibility (Synth), and critical safety metrics—specifically, the presence of toxicophores and Ames mutagenicity scores (Supplementary Table S2). GDCA, GCA, and OAG emerged as the top three candidates. These compounds not only exhibited the strongest predicted binding energies in the cohort (docking scores of −9.5, −9.0, and −8.7 kcal/mol, respectively), but crucially, they possessed the lowest mutagenic risk profiles in the filtered dataset (Ames scores of 0.005, 0.008, and 0.016, respectively). All three compounds exhibited no toxicophores alongside highly favorable Synth scores. This stringent selection rationale successfully filtered out compounds that possessed strong predicted binding energies but higher toxicity risks (e.g., CNP0203565, docking score −8.3 kcal/mol, Ames score 0.120), ensuring that the final chosen lead compounds possess both high therapeutic potential for KIF11 inhibition and exceptional safety margins (Supplementary Table S2).
To gain deeper insights into the specific binding dynamics of these top candidates, detailed analyses of the 2D and 3D interactions within the protein-ligand complexes were conducted (Supplementary Table S3). It was found that the GCA forms hydrogen bonds with the residues LYS111, GLU118, and THR112, and pi–alkyl interactions with the residues PRO27, ARG26, and PHE113. GDCA forms hydrogen bonds with the residues GLY108, PHE113, LYS111, GLY110, THR109, and THR112, and pi–alkyl interactions with the residues LEU132, PRO27, ARG26, and PHE113 (Table 3 and Supplementary Table S3). Further, OAG forms hydrogen bonds with the residues THR112 and GLU118, pi–alkyl interactions with the residue PRO27, pi–pi stacking with PHE113, and an unfavorable negative–negative interaction with GLY110 (Table 3 and Supplementary Table S3).
Binding Profiles and Noncovalent Interactions between the KIF11 Active Site and Selected Natural Compounds

Trajectory analysis using MD simulations
MD simulations were employed to investigate the binding of the top three natural compounds (GCA, GDCA, and OAG) with the KIF11 protein. The simulations provided insights into the conformational dynamics of the ligand–protein complexes.
KIF11 exhibited moderate structural flexibility, with an RMSD fluctuating between 1.5 Å and 4.0 Å, indicating a relatively stable protein structure while retaining some flexibility to accommodate GCA. GCA initially showed an RMSD rise to 5.5 Å before stabilizing, suggesting initial adjustments as GCA searched for and then achieved a stable binding conformation (Fig. 3A). The radius of gyration (rGyr) for GCA remained within a narrow range of 4.8–5.4 Å, further confirming the structural integrity and compactness of GCA throughout the simulation (Fig. 3B). Analysis of RMSF revealed low values for residues within the KIF11 binding site (0.6 Å to 0.8 Å) (Fig. 3E), corroborating the strong ligand-protein interaction. While some flexible regions (higher RMSF values) were observed in the protein, these were primarily located distal to the binding site, minimizing their impact on binding stability. Furthermore, analysis of secondary structure elements (SSE) demonstrated the overall stability of the KIF11 structure, with α-helices and β-strands maintaining approximately 45% SSE content (Fig. 3F). Localized transitions from α-helices to coils suggest minor conformational changes, but GCA consistently interacted with the more stable structural regions of the protein. GCA binding to KIF11 is primarily mediated by hydrogen bonds, forming a strong and specific interaction with key binding site residues (Fig. 3D). Extensive hydrophobic interactions further anchor the ligand within the binding pocket, while ionic interactions and water bridges contribute to the overall stability of the complex (Fig. 3D). RMSF and solvent accessibility surface area (SASA) plot for GCA further reflects stable binding of GCA with KIF11 (Fig. 3C and G). Among the tested complexes, GCA exhibited the most stable binding profile, highlighting its potential as a promising KIF11 inhibitor. These findings suggest that GCA possesses favorable binding characteristics for KIF11 inhibition and warrant further experimental validation and optimization for drug development efforts targeting EC.
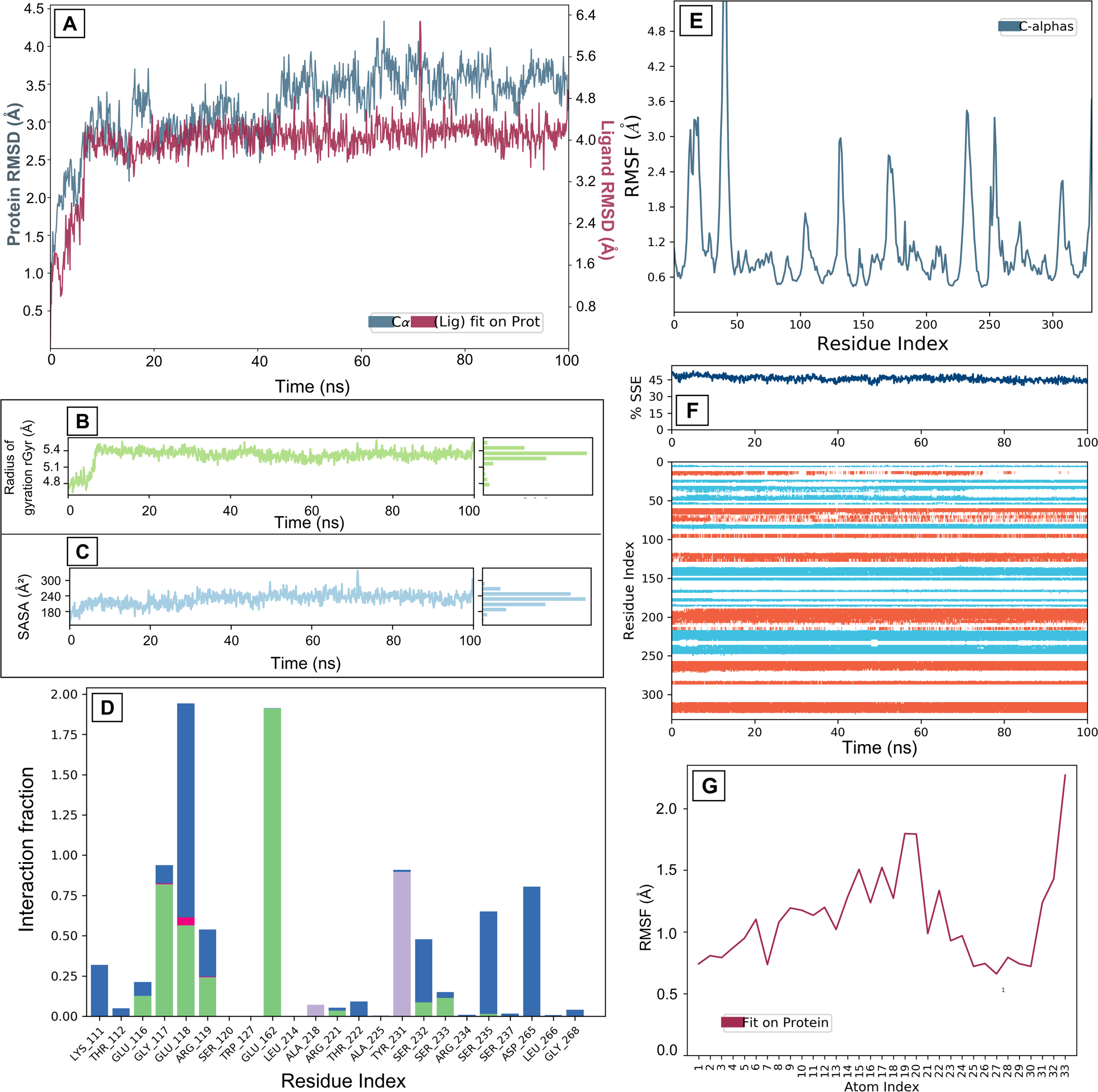
Molecular dynamics interaction graphs for KIF11 and GCA complex:
GDCA binding to the KIF11 protein highlights notable differences in stability and interaction profiles compared to other complexes. The protein’s RMSD fluctuates between 2.4 Å and 3.6 Å, reflecting moderate structural changes and some degree of protein flexibility. The RMSD of GDCA shows initial instability, with fluctuations ranging from 2.9 Å to 9.0 Å, before stabilizing around 4.5 Å (Fig. 4A). This suggests a search phase for a stable binding conformation, followed by a more consistent binding phase. However, RMSF analysis indicates that binding site residues display higher flexibility (0.5–3.8 Å) than observed with GCA (0.6–0.8 Å), suggesting weaker interactions and less stable binding. Prominent peaks in the RMSF graph, particularly near the binding site, further reinforce this observation by indicating increased flexibility that can impact ligand-binding stability (Fig. 4E). The SSEs of the protein during the simulation display overall stability, with consistent presence of α-helices and β-sheets (Fig. 4F). Nonetheless, certain regions exhibit changes over time, with turns and coils demonstrating higher flexibility and less stable structures. These fluctuations point to localized instability that might influence the binding site’s conformation. Interaction analysis reveals that GDCA engages in key stabilizing forces, including hydrogen bonds that maintain specific ligand-residue interactions and hydrophobic interactions that securely accommodate nonpolar regions of GDCA within the binding pocket (Fig. 4D). Ionic interactions, though fewer in number, provide additional electrostatic stabilization, and the presence of water bridges indicates the role of water-mediated interactions in maintaining complex integrity (Fig. 4D). The rGyr for GDCA fluctuates significantly between 4.0 Å and 5.5 Å, indicating substantial conformational changes throughout the simulation (Fig. 4B). This variability in rGyr values suggests that the ligand does not maintain a stable interaction with the receptor and potentially undergoes positional shifts within the binding site or induces structural rearrangements in the complex. Such instability could arise from weaker interactions between the ligand and the receptor, resulting in less consistent compactness and overall structure. Figure 4C and G show RMSF and SASA of GDCA showing more fluctuations than GCA, which depicts a less stable binding than GCA.
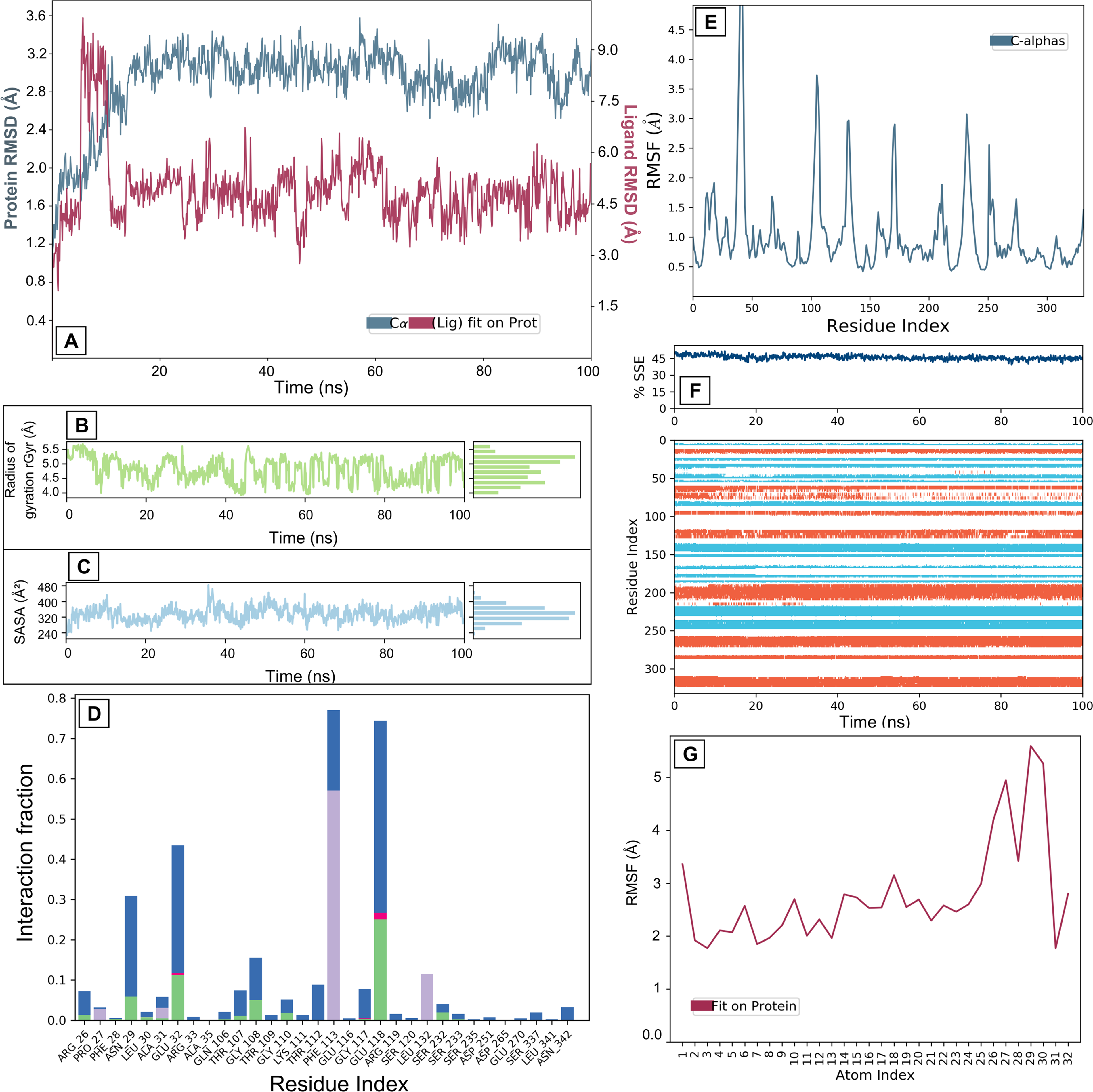
Molecular dynamics interaction graphs for KIF11 and GDCA complex:
MD simulations characterizing the interaction between OAG and KIF11 revealed a dynamic yet moderately stable binding profile. KIF11 exhibited moderate structural fluctuations (RMSD 2.4–2.8 Å), indicating flexibility for ligand adaptation (Fig. 5A). RMSF analysis confirmed moderate flexibility in binding site residues, facilitating optimal ligand accommodation (Fig. 5E). The RMSD of OAG showed an initial stable phase (0–45 ns) at approximately 3.2 Å, a transient increase to 6.4 Å (45–50 ns), and subsequent stabilization between 3.2 Å and 4.0 Å after 50 ns, suggesting an induced-fit mechanism (Fig. 5A). Moderate fluctuations in the rGyr (4.4–5.0 Å) further support conformational flexibility (Fig. 5B). Complex stability was mediated by hydrogen bonds, hydrophobic interactions, ionic interactions, and water bridges (Fig. 5D). The SASA plot shows more fluctuation for OAG than other ligands and same for the RMSF of OAG (Fig. 5C and G). These results suggest moderate ligand-binding stability, balancing robust interactions with dynamic adjustments, supporting the ligand’s potential for therapeutic applications, though further optimization may enhance specificity and reduce variability for more effective KIF11 targeting.
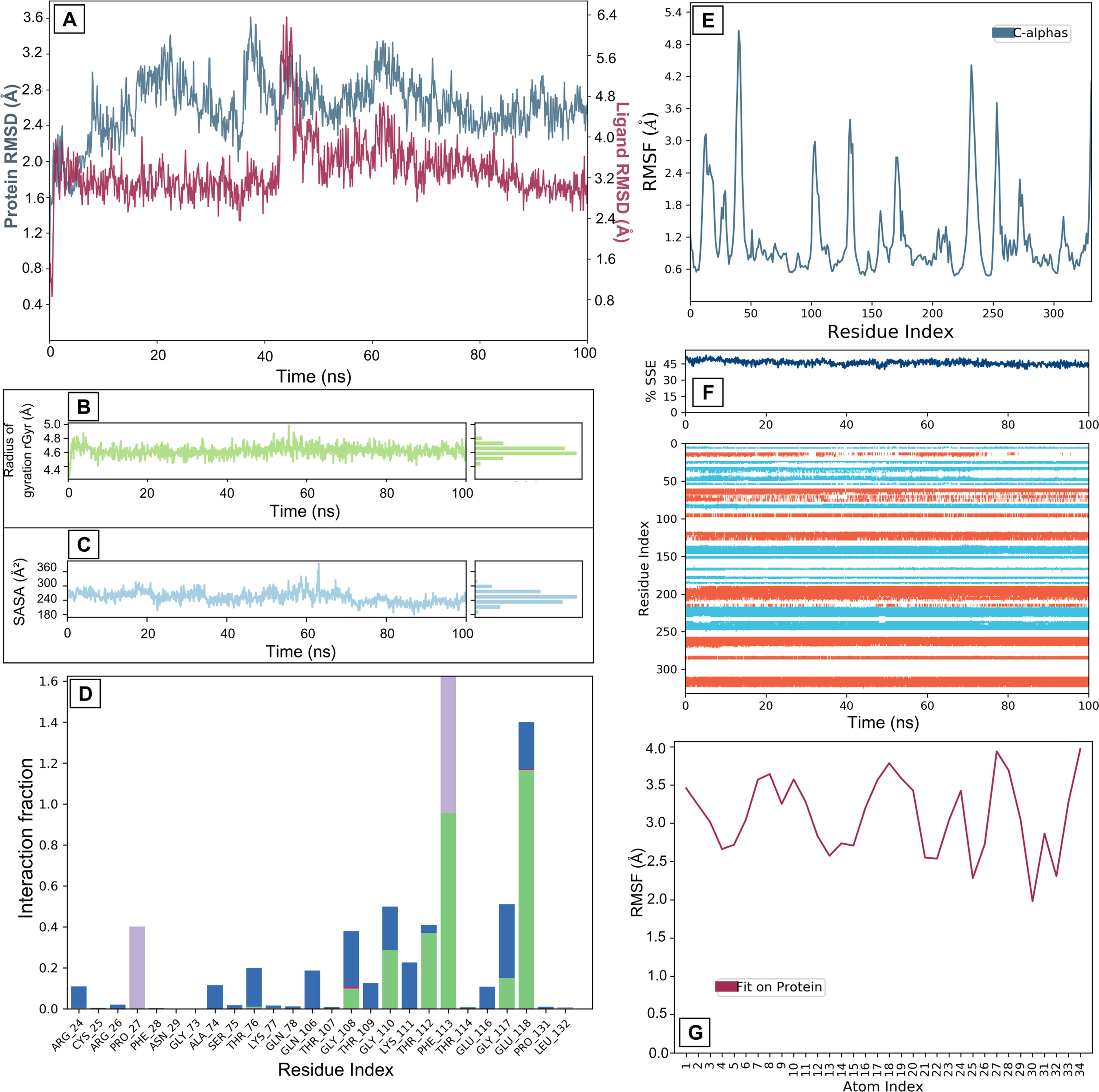
Molecular dynamics interaction graphs for KIF11 and OAG complex:
Discussion
EC, a prevalent gynecological malignancy, particularly among postmenopausal women, develops in the inner lining of the uterus. While previously considered less lethal than other gynecological cancers, its incidence rates have been rising dramatically worldwide, with global cases reaching over 417,000 in 2020 and a significant number of related deaths (Ferlay et al., 2021; Sung et al., 2021). This increase, coupled with the high genetic diversity and heterogeneity of EC, underscores the urgent need for identifying novel and effective therapeutic targets (Hou et al., 2020).
Given its crucial role in cell division and its high expression levels in EC, both at the transcript and protein level (Fig. 1), KIF11, also known as Eg5, was considered a potential therapeutic target for EC. KIF11 is a mitotic motor protein essential for bipolar spindle formation, a process frequently dysregulated in cancer cells (Wang et al., 2025). Its overexpression in various other cancers, including breast, prostate, colorectal, and oral cancers, and its minimal expression in normal, nonproliferating cells, make it an attractive target for selective anticancer therapy (Guo et al., 2022).
To identify potential KIF11 inhibitors, a pharmacophore model was constructed. This model was based on the 3D structure of KIF11 in complex with its native ligand, ADP. The use of a native ligand such as ADP is a common and effective strategy in pharmacophore modeling as it helps to define the key molecular features required for binding within the active site of the target protein (Zhou et al., 2019). This pharmacophore model was then employed for the virtual screening of a natural product library.
This initial screening yielded a substantial number of compounds, which were subsequently filtered down to 56 based on various criteria. Such filtering processes in virtual screening workflows typically involve evaluating drug-likeness, and ADMET properties to prioritize compounds with favorable pharmacokinetic profiles and reduced potential for side effects (Winiwarter et al., 2018). This rigorous filtering ensures that only promising candidates proceed to further stages of analysis, thereby streamlining the drug discovery pipeline (Volkova et al., 2022).
Molecular docking was performed to predict binding modes and affinities of these compounds within the KIF11 active site. Compounds exhibiting favorable binding energies were further evaluated for their pharmacokinetic properties and toxicity profiles. To understand the binding dynamics of KIF11 with potential inhibitors, detailed 2D and 3D interaction analyses were conducted using molecular docking studies with AutoDock Vina (Supplementary Table S3). GDCA exhibited the highest docking score of −9.5 kcal/mol, suggesting a strong binding affinity for the KIF11 active site. GCA and OAG displayed slightly lower docking scores of −9.0 and −8.7 kcal/mol, respectively, indicating slightly weaker interactions (Table 3). These scores provide a quantitative measure of the binding strength between KIF11 and the ligands. GCA formed hydrogen bonds with residues LYS111, GLU118, and THR112, and pi–alkyl interactions with PRO27, ARG26, and PHE113. GDCA engaged in hydrogen bonding with GLY108, PHE113, LYS111, GLY110, THR109, and THR112, along with pi–alkyl interactions with LEU132, PRO27, ARG26, and PHE113. OAG formed hydrogen bonds with THR112 and GLU118, pi–alkyl interactions with PRO27, pi–pi stacking with PHE113, and an unfavorable negative–negative interaction with GLY110, and a pi–alkyl bond with the amino acid PRO27. These specific interactions provide insights into the molecular basis of ligand binding to KIF11 (Table 3 and Supplementary Table S3).
Understanding the dynamic interplay between potential drug molecules and crucial amino acid residues within the active site of the protein is paramount, as these interactions govern the ligand’s inhibitory efficacy. Three promising compounds, exhibiting favorable binding scores and reduced toxicity profiles, were selected for MD simulations to thoroughly investigate their interaction dynamics with KIF11. The MD simulations presented in this study provide valuable insights into the binding dynamics of GCA, GDCA, and OAG to KIF11. Our findings reveal distinct binding profiles for each ligand, highlighting the importance of structural variations in influencing KIF11 interactions.
GCA is a bile acid formed by the conjugation of cholic acid with glycine (Song et al., 2018). It acts as a detergent to solubilize fats for absorption and is itself absorbed (Song et al., 2018). GCA exhibited the most stable binding interaction with KIF11, characterized by a consistent RMSD profile (4.8 Å to 5.6 Å) and persistent hydrogen bonding and hydrophobic interactions (Fig. 3A and D). This stability suggests a strong affinity of GCA for the KIF11 binding site, corroborating its potential as a lead compound for inhibitor development. While previous studies have explored the anticancer properties of bile acids such as GCA, their specific interaction with KIF11 in the context of EC remains relatively unexplored (Wu et al., 2019). Further investigation into the downstream effects of GCA-mediated KIF11 inhibition is warranted. This study identifies GCA and GDCA as potential inhibitors of KIF11. Beyond their established roles as digestive detergents, bile acids are acknowledged as bioactive signaling molecules. Numerous investigations have demonstrated that ursodeoxycholic acid (UDCA) and its conjugates exhibit antitumor properties. Previous research indicated that UDCA can inhibit colon cancer cell proliferation by inducing growth arrest and G2/M phase cell accumulation (Kim et al., 2017). Furthermore, glycoursodeoxycholic acid (GCUDCA) was shown to suppress hepatocellular carcinoma cell proliferation via regulation of the mTOR/S6K1/Rb pathway and induce G1 phase arrest (Zeng et al., 2023). Recently, Li et al. reported that synthetic bile acid derivatives HS-1199 and HS-1200 trigger apoptosis in human cancer cells through p53-independent pathways (Li et al., 2024). These findings provide a foundation for the development of anticancer therapeutics based on bile acid scaffolds.
GDCA is another bile acid derived from deoxycholic acid and glycine (Meessen et al., 2025). It also acts as a detergent to solubilize fats for absorption and is itself absorbed (Meessen et al., 2025). In contrast to GCA, GDCA demonstrated a less stable binding profile, marked by significant fluctuations in its rGyr (4.0–5.5 Å) and higher RMSF values (0.5–3.8 Å) at the binding site (Fig. 4B and E). This suggests a weaker interaction with KIF11, potentially due to structural differences that hinder optimal binding. The observed instability underscores the importance of subtle structural modifications in modulating ligand–protein interactions (Fig. 4A and G). Interestingly, although GDCA demonstrated the highest affinity for KIF11 in static molecular docking, with a docking score of −9.5 kcal/mol, the 100 ns MD simulation revealed that it was less stable than GCA within the protein–ligand complex. This discrepancy highlights the inherent limitations of static docking methodologies, which do not consider the effects of solvent, protein conformational flexibility, and the entropy loss upon binding. Notably, an initial sharp spike in RMSD to approximately 9.0 Å at the outset of the simulation was quickly followed by substantial fluctuations throughout the remaining 100 ns trajectory. Furthermore, the rGyr for GDCA exhibited significant, continuous fluctuations between approximately 4.0 Å and 5.5 Å (Fig. 4B). Coupled with high ligand RMSF peaks approaching 6.0 Å (Fig. 4G), these data indicate that, despite the favorable docking pose of the GDCA-KIF11 complex, the complex lacks long-term structural stability, and the ligand undergoes considerable conformational shifts within the binding pocket. Analyzing these dynamic data facilitates the ranking of lead compounds for subsequent optimization studies.
OAG is a metabolite of androsterone, a steroid hormone (HMDB ID: HMDB0002829) (Wishart et al., 2022) . While the specific role of OAG in EC is not well-established, its interaction with KIF11 warrants further investigation. The binding profile of OAG presented an intermediate level of stability, with moderate RMSD fluctuations and a combination of stabilizing interactions (Figs. 5A–G). Future studies could explore the potential synergistic effects of combining OAG with other KIF11 inhibitors.
This study investigated the binding interactions of three natural compounds—GCA, GDCA, and OAG—with KIF11, a key regulator of mitosis and a promising therapeutic target in EC identified from this research. GCA exhibited the most stable binding profile, suggesting its potential as a lead compound for KIF11 inhibitor development. GDCA showed a slightly weaker interaction, highlighting the impact of structural variations on predicted binding affinity. OAG demonstrated intermediate stability, warranting further investigation into its potential synergistic effects with other inhibitors. These findings underscore the importance of exploring natural compounds as potential KIF11 inhibitors and pave the way for future studies focusing on optimizing their efficacy in targeting EC. Although the in silico pipeline of this study identified GCA as a highly stable hit compound, its biological effects remain to be empirically validated through laboratory experiments. To this end, our forthcoming research will concentrate on the in vitro validation of GCA’s targeted effects, commencing with KIF11 ATPase assays to ascertain whether GCA functions as a direct inhibitor of KIF11’s ATPase activity, which would confirm its binding to the motor protein (DeBonis et al., 2004). Following these biochemical assays, future studies will evaluate the impact of GCA and its analogs on the viability and proliferation of Ishikawa and HEC-1-A cells. Both cell lines are extensively employed as models of EC, exhibiting distinct metabolic and genetic profiles that make them ideal for this validation. Ultimately, these proposed experiments will enable us to assess whether the compounds studied in silico possess translatable therapeutic value in combating EC.
Conclusion
Through a comprehensive computational approach involving pharmacophore-based virtual screening of natural products, molecular docking, and MD simulations, potential KIF11 inhibitors were identified. Notably, GCA emerged as a lead compound, demonstrating the most stable binding interactions with KIF11. While GDCA showed less stable binding, emphasizing the impact of subtle structural differences, OAG presented intermediate stability, warranting further investigation. Ultimately, these findings highlight the significant potential of natural compounds as effective KIF11 inhibitors and pave the way for future drug development efforts aimed at optimizing their efficacy for EC management. To firmly establish these in-silico discoveries, our immediate future directions will prioritize rigorous in vitro experimental validation, including KIF11 ATPase assays and EC cell line proliferation studies, bridging the gap between computational prediction and biological efficacy.
Statements and Declarations
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Authors’ Contributions
S.S.: Methodology, investigation, formal analysis, and writing—original draft. K.M.: Conceptualization, supervision, and writing—review and editing.
Footnotes
Acknowledgments
The authors acknowledge BIT Mesra for providing infrastructure facilities and the Department of Biotechnology (DBT) India (DBT/2022-23/BITR/2078) for Junior Research Fellowship.
Author Disclosure Statement
The authors have stated that they do not have any competing interests.
Funding Information
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.