
Abstract
Statins are widely prescribed lipid-lowering agents that also exert pleiotropic anticancer effects, such as induction of apoptosis and ferroptosis, modulation of autophagy, and remodeling of the tumor microenvironment. Consistent with these multifaceted actions, statins have demonstrated synergistic activity with several chemotherapeutic agents. Emerging evidence indicates that statins modulate the activity of key DNA damage response kinases, such as ataxia-telangiectasia (ATM) and checkpoint kinase 2 (CHK2), in colorectal cancer cells, suggesting a potential impact on DNA repair pathways. To investigate this possibility, publicly available transcriptomic, proteomic, and phosphoproteomic datasets derived from colorectal cancer models treated with atorvastatin or lovastatin were systematically analyzed. Genes and proteins associated with DNA repair exhibiting differential expression, as well as proteins with altered phosphorylation status, were identified. These datasets were subsequently subjected to pathway enrichment and protein-protein interaction network analyses to determine whether statin exposure preferentially affected specific DNA repair pathways. Integrated multi-omics analysis revealed coordinated perturbation of tumor protein p53 (TP53)–centered DNA repair signaling, including pathways involved in TP53 regulation and double-strand break repair. Taken together, these findings suggest that statin-induced alteration of TP53-mediated DNA repair signaling may promote the persistence of DNA damage, thereby increasing the sensitivity of tumor cells to chemotherapy and potentially mitigating resistance mechanisms in colorectal cancer.
Introduction
Statins are competitive inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), the rate-limiting enzyme of the mevalonate pathway, responsible for the biosynthesis of cholesterol and nonsteroidal isoprenoids (Sirtori, 2014). Through this mechanism, statins effectively reduce circulating cholesterol levels and are widely prescribed for the prevention and treatment of cardiovascular disease (Wiggins et al., 2016). Beyond their lipid-lowering effects, there is growing evidence supporting the repurposing of statins as anticancer agents (Lagunas‐Rangel et al., 2024).
Statins have been shown to exert multiple antitumor effects, including the induction of apoptosis and ferroptosis, modulation of autophagy, and reprogramming of the tumor microenvironment toward an antitumor phenotype (Jiang et al., 2021; Lagunas-Rangel, 2025c, 2025a). Their integration into precision cancer medicine strategies is also being explored (Lagunas-Rangel, 2025d). Notably, statins have demonstrated synergistic activity with conventional chemotherapeutic agents and targeted therapies, suggesting their potential to improve treatment efficacy and overcome drug resistance (Jiang et al., 2021; Matusewicz et al., 2020).
Cholesterol is a critical regulator of cell proliferation and cell cycle progression, particularly during the transition to S phase (Singh et al., 2013). Cancer cells also exhibit a greater dependence on cholesterol to maintain optimal mitogenic signaling and survival. This increased demand is reflected in the greater abundance of lipid rafts observed in malignant cells, which serve as platforms for the clustering and activation of oncogenic signaling molecules (Kuzu et al., 2016; Xiao et al., 2023). To meet these requirements, the mevalonate pathway is frequently upregulated in tumors. This pathway not only supplies cholesterol but also generates essential isoprenoid intermediates required for protein prenylation, a post-translational modification necessary for the proper localization and activity of key oncogenic regulators, such as small GTPases (Juarez and Fruman, 2021). In addition to increasing intrinsic cholesterol synthesis, cancer cells actively restructure their microenvironment to secure additional lipid resources. By promoting cholesterol accumulation within the tumor, they contribute to the establishment of an immunosuppressive environment that inhibits the antitumor immune response and facilitates tumor survival and progression (Lagunas-Rangel, 2025b).
On the other hand, colorectal cancer is the third most frequently diagnosed malignancy worldwide and the second leading cause of cancer death (Bray et al., 2024). While therapeutic advances have improved patient outcomes, treatment resistance and incomplete response remain significant clinical challenges (Xie et al., 2020), underscoring the need for complementary therapeutic strategies. A previous study demonstrated that statins modulate the phosphorylation state and activity of multiple kinases in colorectal cancer cells, including key regulators of the DNA damage response, such as ataxia-telangiectasia (ATM) and checkpoint kinase 2 (CHK2) (Lagunas-Rangel et al., 2025). These findings suggest that statins could influence DNA repair networks. To further investigate this possibility, publicly available transcriptomic, proteomic, and phosphoproteomic datasets from colorectal cancer models treated with atorvastatin or lovastatin were systematically analyzed. This comprehensive analysis revealed a coordinated disruption of DNA repair signaling pathways centered on the tumor protein p53 (TP53). Such disruption could promote the persistence of DNA lesions, thereby increasing the susceptibility of tumor cells to chemotherapeutic agents and potentially reducing the development of treatment resistance in colorectal cancer.
Materials and Methods
RNA-seq data analysis
Publicly available RNA sequencing data were obtained from the Gene Expression Omnibus (GEO) with accession number GSE157167 (Norkin et al., 2021). This dataset comprises transcriptomic profiles of colorectal cancer organoids derived from AKP mutant mice with alterations in adenomatous polyposis coli protein (APC), KRAS, and TP53. The organoids were treated for 24 h with vehicle (control), 1 µM atorvastatin, or 0.5 µM lovastatin. Sequencing was performed using the Illumina NextSeq 500 platform. The raw sequencing reads were quality controlled using FastQC (Wingett and Andrews, 2018). To improve data quality, adapter sequences and low-quality bases were trimmed using Trimmomatic (Bolger et al., 2014). The resulting high-quality reads were aligned to the mouse reference genome (GRCm39, Release M37) using HISAT2 (Kim et al., 2019). Gene-level quantification was performed using featureCounts (Liao et al., 2014). To restrict subsequent analyses to genes with robust expression, low-abundance transcripts were filtered out. Specifically, genes with counts per million ≤1 in fewer than two samples per experimental group were excluded. Normalized count data for differential gene expression were analyzed using the edgeR package (Robinson et al., 2010). Genes were classified as significantly upregulated if they showed a log2 fold change (FC) >1.0 with a false discovery rate (FDR) <0.05. Conversely, genes with a log2 FC < −1.0 and an FDR <0.05 were considered significantly downregulated.
Proteomic data analysis
Publicly available proteomic data were retrieved from the study by Nimer et al. (2025), in which HCT-116 colorectal cancer cells were treated with atorvastatin or a vehicle control for 24 h. Cell lysates were prepared in the presence of protease and phosphatase inhibitors. Proteomic profiling was performed using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Peptide separation was carried out on a nanoElute ultra-high-pressure liquid chromatography system coupled to a timsTOF fleX mass spectrometer equipped with a CaptiveSpray nanoelectrospray ion source. Raw LC-MS/MS data were processed using MaxQuant version 2.0.1.0 for protein identification and marker-free quantification (LFQ). Protein abundance was determined based on LFQ intensity values. Differential protein expression between atorvastatin-treated and control samples was assessed using a two-sample Student’s t-test. To account for multiple hypothesis testing, p values were adjusted using the Benjamini–Hochberg for FDR. Proteins with FDR <0.05 were considered statistically significant. The analysis focused specifically on statin-induced alterations in proteins related to DNA repair.
Phosphoproteomic data analysis
Publicly available phosphoproteomic data were obtained from the study by Ouahoud et al. (2021), which characterized global phosphorylation changes in colorectal cancer HCT116 cells following treatment with 2 µM lovastatin or vehicle control for 24 h. The analysis focused specifically on statin-induced alterations in proteins related to DNA repair. Phosphopeptides corresponding to the selected proteins were extracted from the dataset and their quantitative phosphorylation intensity was statistically assessed. For each phosphopeptide, the mean phosphorylation intensity (Σ[x1, x2, …, x9]/n) and the corresponding standard deviation (σ[x1, x2, …, x9]) of the biological replicates for each treatment group were obtained directly from the published dataset. These summary statistics (rather than raw peptide-level intensity values) were used for subsequent comparisons between the lovastatin-treated and control conditions. Consequently, the validity of the inferred significance depends on the accuracy and impartiality of the original estimates. The presence of negative values is due to the normalization and calibration procedures used in the original dataset. Specifically, phosphorylation intensities were quantified relative to the reference peptides used for instrument calibration and quality control. Therefore, the negative values reflect protein phosphorylation levels lower than those of the reference peptides, rather than actual negative intensities. Furthermore, mixed peptides lacking serine, threonine, or tyrosine residues were included as negative controls, while mixed kinase substrate peptides were used as positive controls to validate the assay’s performance. Differences in phosphorylation levels between treatment groups were assessed using Welch’s t-test for independent samples. Degrees of freedom were estimated using the Welch–Satterthwaite approximation, and a p-value was calculated for each phosphosite. To correct for multiple hypothesis tests, p values were adjusted using FDR correction. Proteins were considered to have statistically significant changes in phosphorylation when the adjusted FDR was ≤0.05.
Enrichment analysis and functional network assessment
Genes and proteins identified as significantly downregulated in the RNA sequencing together with proteins showing reduced phosphorylation at activating regulatory sites (consistent with decreased protein activity), were subjected to pathway enrichment analysis to determine which specific DNA repair pathways were overrepresented. Pathway enrichment analysis was performed using the Reactome web platform (Joshi-Tope, 2004) with default parameters. To further investigate the potential functional relationships between the identified proteins, protein–protein interaction networks were constructed using the STRING database (Szklarczyk et al., 2021). STRING was used to assess known and predicted functional associations, facilitating network visualization and the identification of biologically connected groups.
Results
Atorvastatin and lovastatin alter the transcriptomic expression of a subset of DNA repair proteins in colorectal cancer cells
To assess the impact of statin treatment on genes associated with DNA repair, RNA sequencing data from colorectal cancer organoids derived from AKP mutant mice treated with atorvastatin or lovastatin were analyzed. By restricting the analysis to genes involved in DNA repair processes, only a limited subset showed significant differential expression (Table 1). Specifically, four genes exhibited significantly decreased expression after lovastatin treatment (Trp53, Cdkn1a, Rbbp4, and Pten), while only one gene showed significantly decreased expression in response to atorvastatin (Trp53). Notably, TP53 expression was reduced in both treatment conditions, suggesting a possible shared effect of these statins. No significant alterations in the expression of DNA repair genes were observed in wild-type colorectal cells following treatment with atorvastatin or lovastatin, compared with vehicle-treated controls.
DNA Repair–Associated Genes That Show Differential Expression after Treatment with Atorvastatin or Lovastatin Compared with Vehicle Control

To better characterize the effects of statins, publicly available proteomic data from colorectal cancer cells treated with atorvastatin were analyzed. Although a small subset of proteins related to DNA repair showed a modest increase in their abundance that was statistically significant at the nominal p-value level, this significance did not hold after adjustment for multiple tests, indicating that these changes did not remain significant when controlling for the FDR (Table 2).
DNA Repair–Associated Proteins That Show Changes in Abundance after Treatment with Atorvastatin

Lovastatin induces phosphorylation changes in a subset of DNA repair proteins in colorectal cancer cells
Since the activity of many DNA repair proteins is regulated by post-translational modifications, particularly phosphorylation, publicly available phosphoproteomic data from colorectal cancer cells treated with lovastatin were analyzed (Table 3). This analysis identified nine DNA repair-associated proteins that exhibited significant changes in phosphorylation status (breast cancer type 1 susceptibility protein [BRCA1], Fanconi anemia group A protein [FANCA], ATM, CHK2, retinoblastoma-binding protein 8 [RBBP8], TP53, tumor protein p73 (TP73), double minute 2 protein (MDM2), and Phosphatase and Tensin Homolog (PTEN)), four of which showed alterations at multiple phosphosites. Fanconi anemia group D2 protein (FANCD2) exhibited a trend toward reduced phosphorylation; however, this change did not reach statistical significance (FDR = 0.59). In total, 19 phosphopeptides were significantly affected: 13 showed reduced phosphorylation levels after lovastatin treatment compared with the control group, while 6 showed increased phosphorylation. Notably, most of the phosphorylation changes occurred at regulatory sites previously associated with modulating protein activity or stability. These alterations are consistent with reduced enzyme activity or decreased protein stability, which could contribute to the functional impairment of the affected DNA repair proteins.
DNA Repair–Associated Proteins That Show Significantly Altered Phosphorylation Levels after Lovastatin Treatment
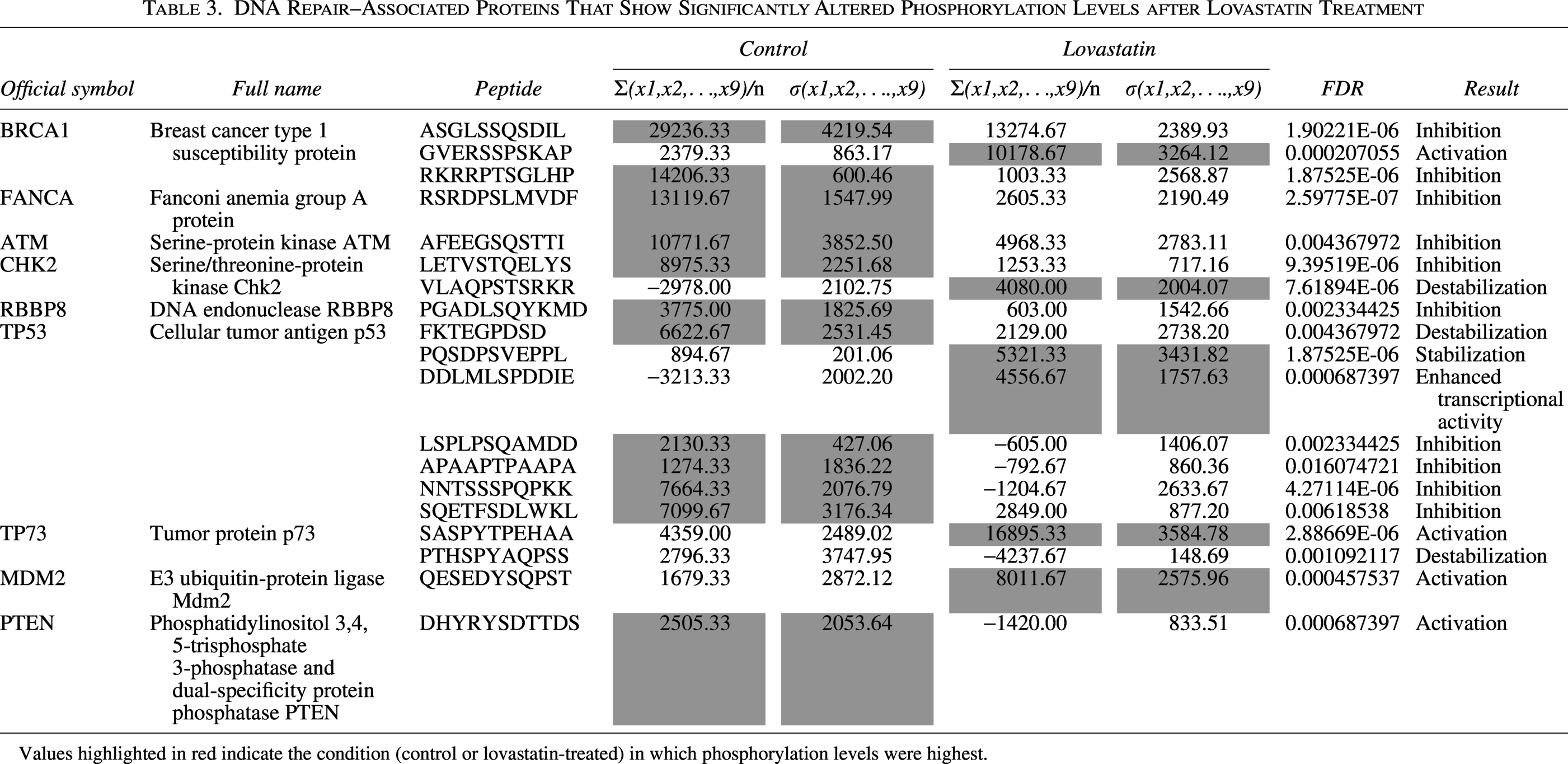
Values highlighted in red indicate the condition (control or lovastatin-treated) in which phosphorylation levels were highest.
Enrichment and network analyses suggest disruption of TP53-mediated DNA repair signaling by statins
To determine whether specific DNA repair pathways were preferentially affected by statin treatment in colorectal cancer cells, a pathway enrichment analysis was performed using DNA repair-associated genes and proteins that exhibited reduced phosphorylation at activating regulatory sites consistent with decreased protein activity. The analysis revealed a significant enrichment of pathways centered on TP53 signaling and responses to DNA damage, including the regulation of TP53 activity through phosphorylation (FDR = 8.18 × 10−10), regulation of TP53 activity (FDR = 2.15 × 10−10), regulation of TP53 activity through methylation (FDR = 8.08 × 10−8), regulation of TP53 degradation (FDR = 4.67 × 10−7), transcriptional regulation by TP53 (FDR = 2.15 × 10−10), and recruitment and ATM-mediated phosphorylation of repair and signaling proteins at DNA double-strand breaks (FDR = 1.80 × 10−6).
Taken together, these results suggest a coordinated disruption of TP53-centered DNA repair signaling pathways following statin treatment (Fig. 1A). Consistent with this finding, protein–protein interaction network analysis identified TP53 as a central node within the affected network, further supporting the disruption of TP53-mediated DNA repair signaling (Fig. 1B).
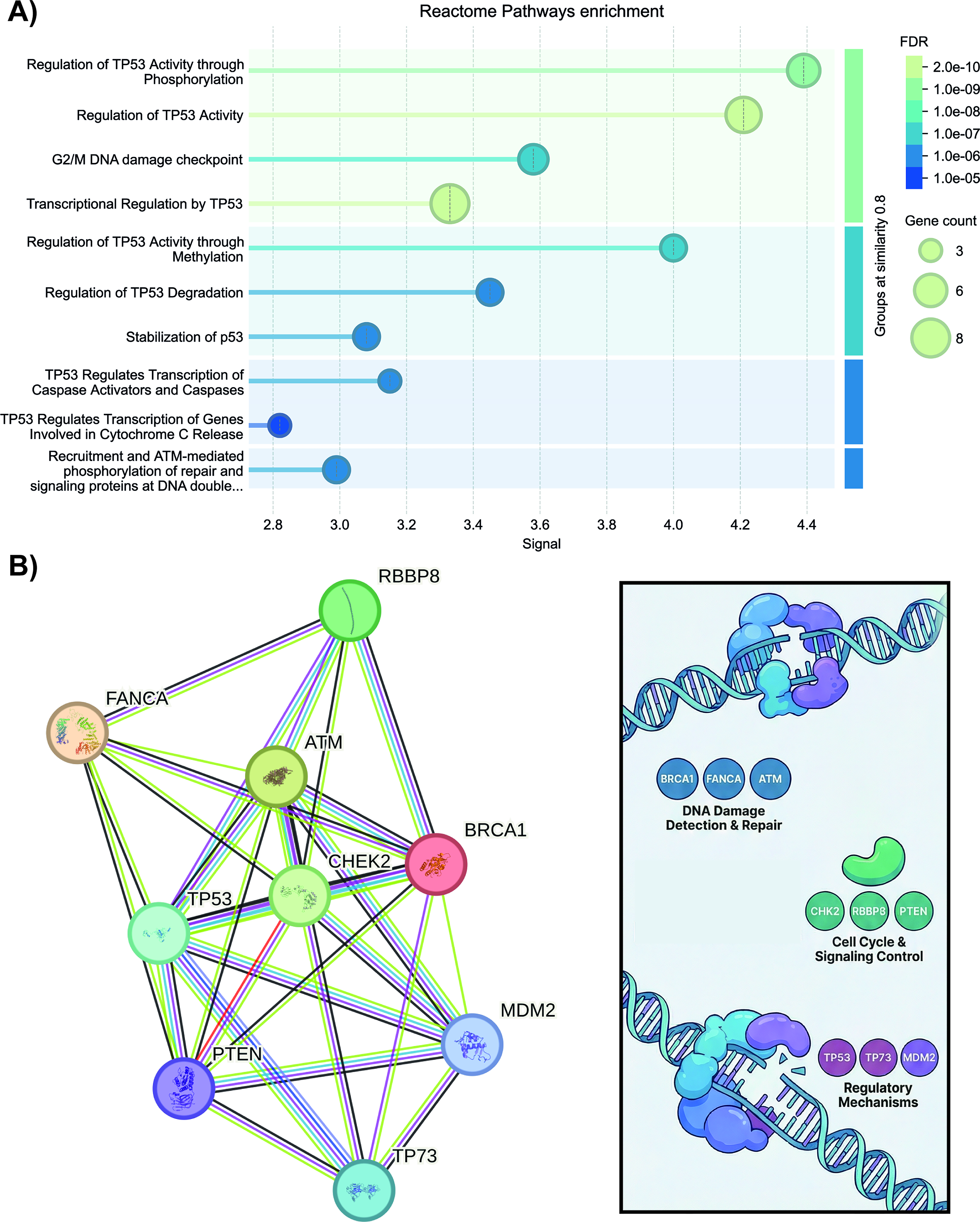
Enrichment analysis and interaction networks of DNA repair proteins affected by statins.
Discussion
Most chemotherapeutic agents exert their anticancer effects by inducing DNA damage, which ultimately triggers cell death in rapidly proliferating tumor cells. However, in response to genotoxic stress, cancer cells frequently upregulate DNA repair pathways, enhancing their survival and contributing to therapeutic resistance (Groelly et al., 2023). In this context, statins have emerged as potential sensitizing agents capable of disrupting these adaptive resistance mechanisms. Several studies have reported synergistic effects when statins are combined with conventional chemotherapeutic agents, such as 5-fluorouracil (Xing et al., 2024), doxorubicin (Buranrat et al., 2017), cyclophosphamide (Dewidar et al., 2023), and pentoxifylline (Castellanos-Esparza et al., 2018). Similar synergistic interactions have also been observed with tyrosine kinase inhibitors such as sorafenib (Cheng et al., 2017), vemurafenib (van Leeuwen et al., 2022), and gefitinib (Cemeus et al., 2008).
While the evidence presented in this study is based on the reanalysis of third-party datasets generated under heterogeneous experimental conditions—including the use of different cell lines (murine and human) with diverse TP53 backgrounds (wild-type and mutant), different statins (atorvastatin and lovastatin), and varying treatment durations—this approach remains highly informative. It serves as a scientific compass, leveraging large-scale data to guide and refine future experimental research. The findings suggest a possible association between statin treatment and alterations in TP53-related DNA repair pathways. In this context, the data suggest that the improved therapeutic response observed in vitro after statin exposure could be mediated, at least in part, by the disruption of TP53-dependent DNA repair signaling. Importantly, these observations should be interpreted with caution and considered preliminary, not conclusive.
Atorvastatin and lovastatin, the statins used in the transcriptomic study, share a common mechanism of action, the inhibition of HMGCR, which leads to the suppression of the mevalonate pathway. Despite sharing a common mechanism of action, atorvastatin and lovastatin differ in their origin and physicochemical properties. Lovastatin is a naturally occurring fungal metabolite, while atorvastatin is entirely synthetic. Both compounds are generally classified as lipophilic statins; however, their effective lipophilicity in biological systems differs. Atorvastatin exists predominantly in its ionized (active acid) form under physiological conditions, which reduces its membrane permeability relative to its nominal logP value. In contrast, lovastatin is administered as a more nonpolar, neutral lactone prodrug, allowing for greater passive diffusion across cell membranes. Notably, the lipophilic nature of both statins facilitates passive diffusion across cell membranes, promoting not only hepatic uptake but also distribution to extrahepatic tissues (Lagunas-Rangel, 2025d). These properties could contribute to the more extensive transcriptional changes observed with lovastatin in the RNA sequencing dataset.
It is worth noting that no transcriptional changes in TP53 expression were observed in wild-type colorectal cells, suggesting that its regulation may be related to the presence of mutations in APC, KRAS, and TP53, which are commonly found in colorectal cancer. Previous studies have shown that statins promote the degradation of mutant TP53 (Wang et al., 2025). Mechanistically, mutant p53 stability is maintained by histone deacetylase 6/heat shock protein 90-dependent chaperone activity, supported by RAS homolog family member A (RhoA) geranylgeranylation. By inhibiting the mevalonate pathway, statins disrupt these processes, thereby promoting the destabilization and degradation of mutant p53 (Ingallina et al., 2018). Furthermore, proteomic analysis performed on HCT-116 cells with wild-type TP53 revealed no changes in TP53 protein levels. Although the transcriptomic (murine cells with mutant Trp53) and proteomic analyses (human cells with wild-type TP53) were conducted in different models, the observed differences could be related to the TP53 status. In this context, it is possible that TP53-associated effects are more pronounced in mutant environments, which could indicate a phenotype potentially more sensitive to the combined action of statins and chemotherapeutic agents. However, this interpretation remains speculative and will require further research for confirmation.
This study also observed that statin treatment not only reduces mutant TP53 expression levels but also decreases phosphorylation at multiple residues of the wild-type protein. Since TP53 phosphorylation is crucial for its stabilization and transcriptional activity, these alterations could affect its functional capacity, including its role in regulating DNA repair signaling pathways.
In addition to its well-established role in coordinating cell cycle arrest in response to DNA damage, TP53 also plays a direct and multifaceted role in regulating DNA repair processes. Besides functioning as a transcription factor that controls the expression of repair-related genes, TP53 is actively involved in multiple DNA repair pathways. These include nucleotide excision repair, base excision repair, mismatch repair, homologous recombination (HR), and nonhomologous end joining (Williams and Schumacher, 2016). When DNA damage occurs, ATM is activated at double-strand breaks and phosphorylates TP53 directly or via CHK2, leading to the stabilization of TP53 and the transcriptional activation of repair-associated genes, such as cyclin-dependent kinase inhibitor 1 (CDKN1A or p21), which reinforces cell cycle arrest to allow time for repair (Vogelstein et al., 2000). MDM2 forms a negative feedback loop with TP53 by ubiquitinating and degrading it, thereby adjusting the DNA damage response amplitude (Ellison et al., 2025). TP73 is a member of the p53 family that cooperates with TP53 in the induction of genes involved in DNA repair and apoptosis (Bourdon, 2007). Phosphatase and Tensin Homolog (PTEN) supports TP53 function indirectly by suppressing phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling, which otherwise promotes MDM2-mediated TP53 degradation, and also contributes to homologous recombination (HR) and chromosomal stability (Ming and He, 2012). In the execution of DNA repair, BRCA1 and RBBP8 (also known as CtIP) promote DNA end resection and HR, processes that TP53 functionally coordinates to maintain genomic integrity (Mozaffari et al., 2021; Yoshida and Miki, 2004). FANCA supports HR through the Fanconi anemia pathway in the repair of cross-links between chains (Benitez et al., 2018). Together, these proteins form a coordinated network in which TP53 acts as the central decision-maker, integrating kinase signaling, chromatin dynamics, nucleotide supply, and choice of repair pathway to preserve genomic integrity.
Some experimental observations may support the findings of this study. In HCT116 cells, lovastatin has been reported to induce phosphorylation of γH2AX, a commonly used marker of DNA damage, at micromolar concentrations (5–15 µM) (Huang et al., 2024). Furthermore, treatment with simvastatin (2–20 µM) in combination with ionizing radiation (IR) has been associated with a delay in the repair of IR-induced DNA damage in the same cell line (Lee et al., 2017). In a related context, simvastatin and pravastatin (2 µM) have been shown to attenuate the TP53 response after DNA damage with 5-fluorouracil (20 µM) (Pääjärvi et al., 2005).
In conclusion, this integrative multiomics analysis suggests that statins are associated with coordinated alterations in TP53-related DNA damage response pathways in colorectal cancer models. These changes are observed primarily at the level of gene expression and protein phosphorylation. While these findings are consistent with a possible modulatory effect of statins on TP53-dependent signaling, they remain correlative and do not establish a direct functional alteration of DNA repair capacity. Therefore, the proposed role of statins in sensitizing tumor cells to genotoxic therapies should be considered a hypothesis requiring further experimental validation. Overall, this study provides a framework for understanding how statins may influence DNA damage response pathways and supports future research on their potential use as adjuvant agents in colorectal cancer therapy.
This study has several limitations that should be acknowledged. First, the analyses relied exclusively on publicly available multiomics datasets generated under different experimental conditions, which could introduce heterogeneity related to cell line characteristics, treatment duration, and analytical platforms. Second, only atorvastatin and lovastatin were examined, limiting the generalizability of these findings to other statins with different pharmacological properties. Third, the work is based on in vitro models of colorectal cancer that do not fully capture the complexity of the tumor microenvironment or the systemic influences present in vivo. Furthermore, the observed molecular alterations are primarily correlative and require functional validation to establish direct mechanistic links. Despite these limitations, the integrative analysis provides significant information on the potential impact of statins on TP53-centered DNA repair signaling. These findings offer a mechanistic framework to guide future experimental validation and support further research on statins as adjuvant agents to enhance response to chemotherapy and counter resistance in colorectal cancer.
Footnotes
Author’s Contributions
F.A.L.R. contributed to conceptualization, data curation, formal analysis, investigation, methodology, resources, validation, visualization, writing of the original draft, and writing, review, and editing of the article.
Availability of Data and Materials
Generative AI Statement
AI-assisted tools were employed exclusively for linguistic editing and stylistic refinement. All concepts, data, analyses, interpretations, and conclusions are original and were developed independently by the author.
Author Disclosure Statement
The author declares no conflict of interest.
Funding Information
This research did not receive any specific grant from funding agencies in the public, commercial, or not‐for‐profit sectors.