
Abstract
Chronic renal failure (CRF) is a major risk factor for acute myocardial infarction (AMI) progression and poor outcomes, but the molecular, microbial, and metabolic features of AMI combined with CRF remain unclear. In this study, four Gene Expression Omnibus datasets were analyzed separately. GSE194388 and GSE180393 were used as discovery cohorts for AMI and CRF, and GSE109048 and GSE142153 were used as validation cohorts. Fecal samples from CRF patients without AMI (negative control [NC], n = 10) and patients with AMI and advanced CKD (SY, n = 10) were analyzed by 16S rRNA sequencing. Matched serum samples were analyzed by untargeted metabolomics. Four shared candidate hub genes, PTPRC, ITGAL, CD44, and SELL, were identified and showed preliminary discriminatory performance in validation datasets, although potential overfitting should be considered. These genes were positively correlated with increased immune cell subsets. Compared with NC, SY showed exploratory taxa-level microbiota differences, including increased Enterobacterales and decreased Bifidobacterium, without significant global diversity differences. Differential serum metabolites, including increased
Keywords
Introduction
The heart and kidneys interact closely, and dysfunction in one organ can aggravate injury in the other. Chronic renal failure (CRF) is an important risk factor for cardiovascular disease progression. The cardiovascular mortality of hemodialysis patients with renal failure is reported to be 10–30 times higher than that of the general population (Geng et al., 2024). In acute coronary syndrome, patients with CRF have a higher risk of recurrent acute myocardial infarction (AMI), heart failure, and atypical angina symptoms than patients without CRF (Moukarbel et al., 2014). CRF-related cardiac injury is partly associated with the accumulation of toxic metabolites, such as creatinine (Brankovic et al., 2020; van Veldhuisen et al., 2016). However, other metabolites and molecular changes may also contribute to myocardial injury in patients with CRF. Therefore, identifying molecular and metabolic features in AMI patients with CRF may help clarify the pathological basis of cardiorenal syndrome.
The gut microbiota also participates in cardiorenal interactions. Gut microorganisms regulate immunity, barrier integrity, and glucose and lipid metabolism (Adak and Khan, 2019). During CRF, gut microbiota dysbiosis, intestinal barrier impairment, and bacterial translocation may occur (Wang et al., 2020). Microbiota-derived uremic toxins, including indoxyl sulfate and p-cresyl sulfate, can activate inflammatory responses and promote renal fibrosis (Headley et al., 2025). Gut microbiota has also been linked to cardiovascular disease and AMI (Chen et al., 2025). For example, reduced probiotics may increase trimethylamine metabolism and the production of trimethylamine N-oxide, which has been associated with ventricular remodeling, reduced cardiac function, and myocardial fibrosis (de Vos et al., 2022; Witkowski et al., 2020). In addition, gut barrier injury may induce systemic inflammation and further aggravate myocardial damage (Yu et al., 2025). Thus, profiling gut microbiota may provide additional information on the pathological pathway of AMI combined with CRF.
Transcriptomics provides another approach to characterize complex disease phenotypes (Chen et al., 2024). Previous AMI studies have identified differentially expressed genes (DEGs) related to endothelial ferroptosis and macrophage apoptosis (Xie et al., 2024). Clinical transcriptomic profiling of peripheral blood mononuclear cells from patients with renal injury has also identified prognostic biomarkers and pathways related to hypoxia, metabolic adaptation, inflammation, renal protection, and oxidative stress (Normahani et al., 2022). In addition, worsening renal function after AMI is common and is associated with adverse prognosis, further supporting the close interaction between myocardial injury and renal dysfunction (Jin et al., 2023). However, the molecular features of AMI combined with CRF remain incompletely understood.
In this study, we integrated transcriptomics, 16S rRNA sequencing, and untargeted metabolomics to explore candidate molecular, microbial, and metabolic features associated with AMI combined with CRF. Public Gene Expression Omnibus (GEO) datasets were used to identify shared immune-related hub genes in AMI and CRF. Clinical fecal and serum samples were then used to assess gut microbiota and serum metabolite differences between CRF patients without AMI and patients with AMI and advanced CKD. Finally, microbiota–metabolite correlations were analyzed to generate hypotheses for future validation.
Methods
Data collection
AMI-related datasets GSE194388 and GSE109048, and CRF-related datasets GSE180393 and GSE142153 were downloaded from the GEO database. GSE194388, including 5 AMI samples and 5 normal controls, and GSE180393, including 53 CRF samples and 9 normal controls, were used as independent discovery datasets. GSE109048, including 19 AMI samples and 19 normal controls, and GSE142153, including 20 CRF samples and 10 normal controls, were used only as external validation datasets for candidate hub genes. Because these datasets came from different studies, they were analyzed separately and were not merged into one expression matrix.
DEG analysis and functional enrichment
Differential expression analysis was performed separately in GSE194388 and GSE180393 using the limma package in R. No cross-dataset normalization or batch-effect correction was applied because datasets were not combined for DEG calling. DEGs were identified within each dataset, and genes with concordant expression changes in both datasets were retained for downstream analysis. Genes with p < 0.05 and |logFC| >1 were considered candidate DEGs. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed using clusterProfiler. Pathways with adjusted p values <0.05 were considered enriched.
PPI analysis and hub gene assessment
The shared DEGs were imported into STRING 11.0, with species set as humans, to construct a protein–protein interaction (PPI) network. Cytoscape 3.9.1 and the MCODE algorithm were used for module analysis. The MCC, DMNC, DNC, Gradient, and EPC algorithms in CytoHubba were used to select the top five hub genes from each algorithm. The intersecting hub genes were retained, and GeneMANIA was used to visualize their interaction network. Candidate hub genes were further evaluated in GSE109048 and GSE142153 by receiver operating characteristic (ROC) analysis using the pROC package. The area under the curve (AUC) was calculated. This analysis was considered exploratory because of the small discovery cohort and the lack of prospective validation.
Immune infiltration analysis
Immune infiltration was estimated in GSE194388 and GSE180393 using the CIBERSORT algorithm in the IOBR package with the LM22 immune cell matrix. The relative abundance of 22 immune cell types was calculated for each sample. Group differences between AMI and controls and between CRF and controls were analyzed. Spearman correlation was used to assess relationships between hub genes and immune cell abundance, and ggplot2 was used for visualization.
Participants and sample collection
Clinical omics analysis included 20 participants: CRF patients without AMI as the negative control (NC) group (n = 10) and patients with AMI and advanced CKD as the SY group (n = 10). Fecal samples from all participants were used for 16S rRNA sequencing, and matched serum samples were used for untargeted metabolomics. Microbiota–metabolite correlation analysis was performed in paired fecal and serum samples.
The SY group met AMI diagnostic criteria, including chest pain with dynamic cardiac troponin I elevation and ST-T changes, imaging evidence of new myocardial loss or regional wall motion abnormality, or angiographically confirmed coronary thrombus. The NC group included CRF patients without AMI or myocardial injury within the previous 3 months based on cardiovascular evaluation. Exclusion criteria included type 2 myocardial infarction secondary to infection, heart failure, or anemia; severe cardiac complications; obstructive nephropathy; renal insufficiency aggravated by previous percutaneous coronary intervention; severe infection or sepsis; drug abuse; pregnancy; and lactation. Peripheral venous blood and fecal samples were collected before coronary angiography or temporary dialysis. Serum was separated by centrifugation, and all samples were stored at −80°C. The study was approved by the Ethics Committee of Hunan Aerospace Hospital (HTYY2023LLSH-068-01), and all participants provided written informed consent.
16S rRNA sequencing and microbiota analysis
Fecal DNA was extracted using the E.Z.N.A. Stool DNA Kit. PCR products were purified with AMPure XP beads, quantified using a Qubit fluorometer, and assessed with an Agilent 2100 Bioanalyzer before sequencing on the NovaSeq PE250 platform. Operational taxonomic units were clustered using UPARSE at 97% similarity. Alpha diversity was assessed using Chao1 and Shannon indices, and beta diversity was evaluated using weighted UniFrac principal coordinates analysis in QIIME. LEfSe analysis was performed using p < 0.05 and LDA score ≥4. Random forest analysis and ROC curves were used as exploratory approaches to assess taxa-level discriminatory signals. Because of the small cohort and the absence of significant global diversity differences, microbiome findings were interpreted as exploratory.
Untargeted metabolomics and data processing
Serum samples were mixed with
Correlation analysis and statistics
Pearson correlation and two-way orthogonal PLS (O2PLS) were used to examine relationships between differential taxa and serum metabolites. Features with the top 10 joint loadings were selected for visualization. Data were presented as mean ± standard deviation. SPSS 21.0 was used for statistical analysis. Independent-sample t tests were used for normally distributed data, and rank-sum tests were used for nonnormally distributed data. p < 0.05 was considered statistically significant.
Results
Identification of candidate hub genes shared by AMI and CRF
Independent DEG screening in the discovery datasets GSE194388 (AMI) and GSE180393 (CRF) identified 460 shared genes with concordant expression trends across the two disease settings (Fig. 1A–C; top DEGs listed in Table 1). Functional enrichment analysis of these shared genes revealed a predominant convergence on leukocyte activation, cell adhesion, and immune response regulation. Specifically, GO and KEGG analyses highlighted pathways involved in cytokine signaling and structural components facilitating cell migration (Fig. 1D, E). While multiple “viral infection” pathways were enriched, these signatures reflect the activation of conserved innate immune and interferon-mediated modules characteristic of sterile inflammation in AMI and CRF, rather than active viral pathogenesis.
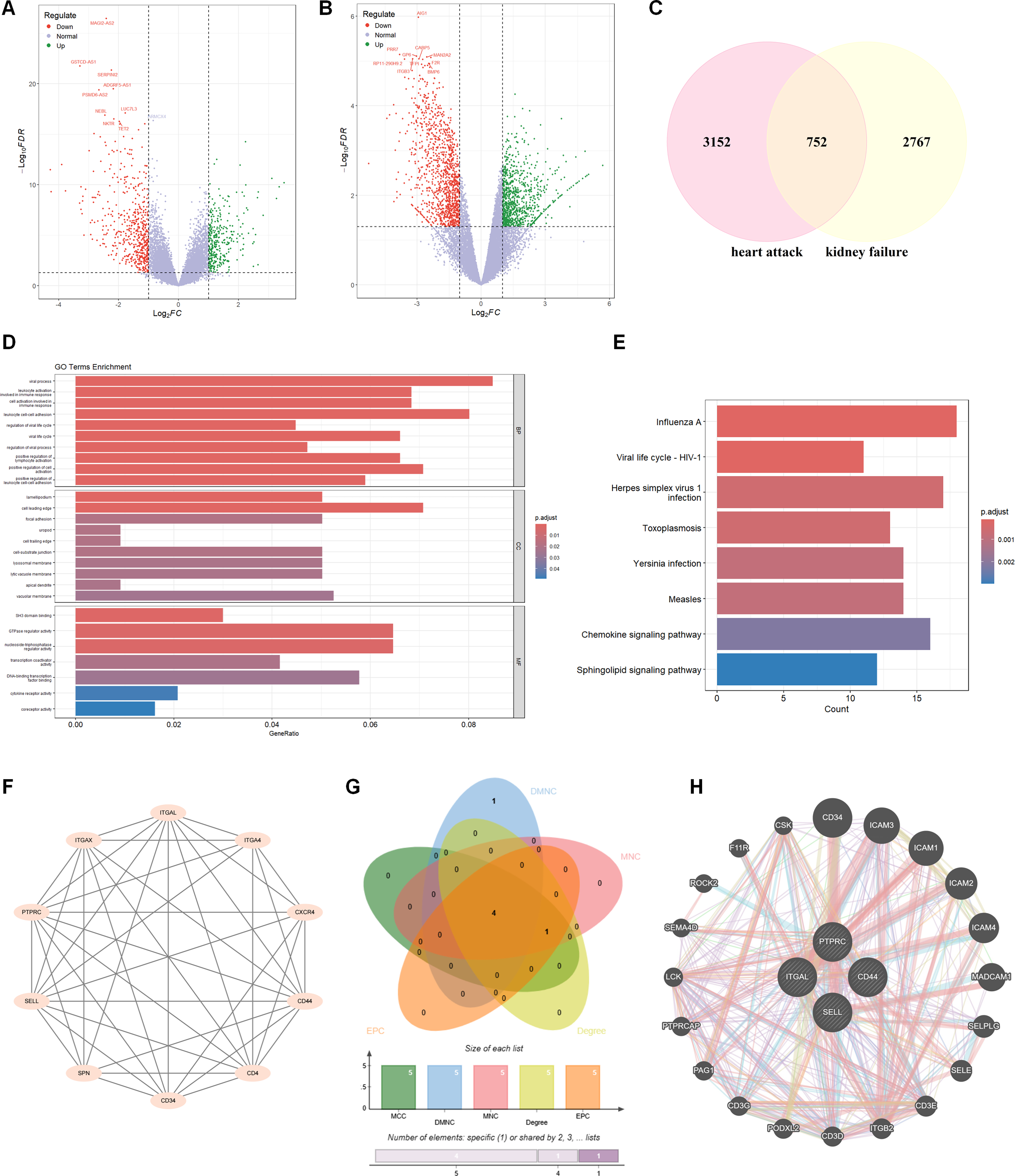
Screening of candidate hub genes shared by the AMI and CRF discovery datasets. The volcano plot of DEGs between AMI patients and normal controls
Top Significantly Downregulated Genes in Acute Myocardial Infarction (GSE194388) and Chronic Renal Failure (GSE180393) Datasets
To pinpoint the drivers of this inflammatory network, topological analysis of the PPI landscape was performed using MCODE and multiple centrality algorithms. This screening strategy prioritized four candidate hub genes, namely, PTPRC, ITGAL, CD44, and SELL (Fig. 1F–H). All four genes encode key cell surface molecules involved in leukocyte trafficking and adhesion, suggesting that dysregulated immune cell migration may represent a shared immune-related feature linking AMI and CRF.
Preliminary discriminatory performance of the candidate four-gene signature and its association with immune infiltration
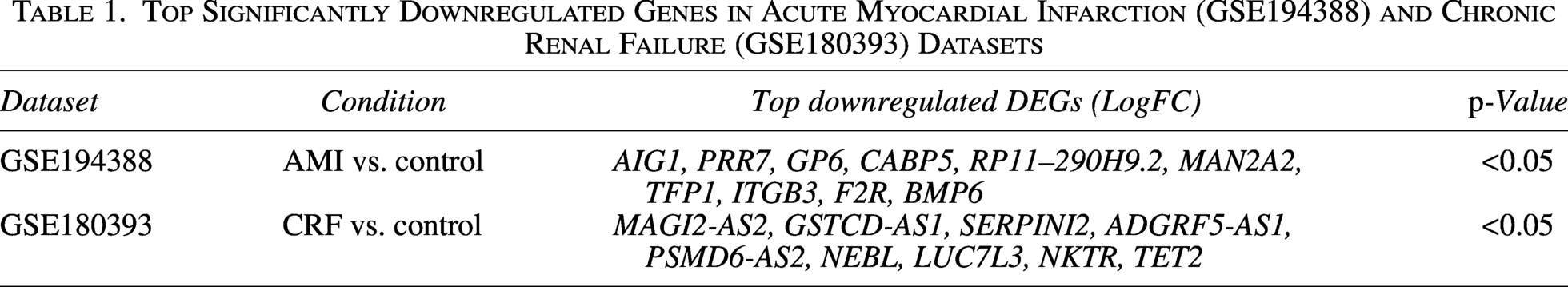
Before examining the association between the candidate hub genes and immune infiltration, we performed a preliminary evaluation of the four-gene signature in the independent GEO validation datasets. In GSE109048, the four-gene signature yielded an AUC of 0.941 for distinguishing AMI samples from controls (Fig. 2A). In GSE142153, the corresponding AUC for distinguishing CRF samples from controls was 0.986 (Fig. 2B). Although these findings suggest potentially favorable discriminatory ability, they should be interpreted with caution because the discovery-stage feature selection was based on small public datasets, and no truly independent clinical external cohort, resampling-based validation, or prospective validation was available in the present study. Therefore, these ROC results should be regarded as preliminary rather than definitive evidence of diagnostic performance. After that, immune infiltration in the GSE194388 and GSE180393 datasets was analyzed. It revealed that, compared with the healthy, the estimated proportion of neutrophils and T cells-CD4 memory resting was significantly increased (p < 0.05) in AMI, and the estimated proportion of T cells-CD8 was significantly decreased (p < 0.01) in AMI (Fig. 2C). Compared with the healthy, the estimated proportion of monocytes was significantly increased (p < 0.01), and the estimated proportion of CD4 memory resting was significantly decreased (p < 0.01) in CRF (Fig. 2D). In addition, the correlation between the four candidate hub genes and immune infiltration was analyzed. As shown in Figure 2E, the four candidate hub genes, PTPRC, ITGAL, CD44, and SELL, were positively correlated with immune infiltration in the GSE194388 dataset, among which ITGAL shows the strongest correlation with immune infiltration. As presented in Figure 2F, the four candidate hub genes, PTPRC, ITGAL, CD44, and SELL, were positively correlated with immune infiltration in the GSE180393 dataset, among which PTPRC showed the strongest correlation with immune infiltration. The above results indicated that the four candidate hub genes were positively correlated with the infiltration of multiple immune cell subsets.
Preliminary discriminatory performance of the candidate four-gene signature and its association with immune infiltration. ROC curves of the four-gene signature in the GEO validation datasets for AMI
Changes in the gut microbiota of AMI patients with CRF
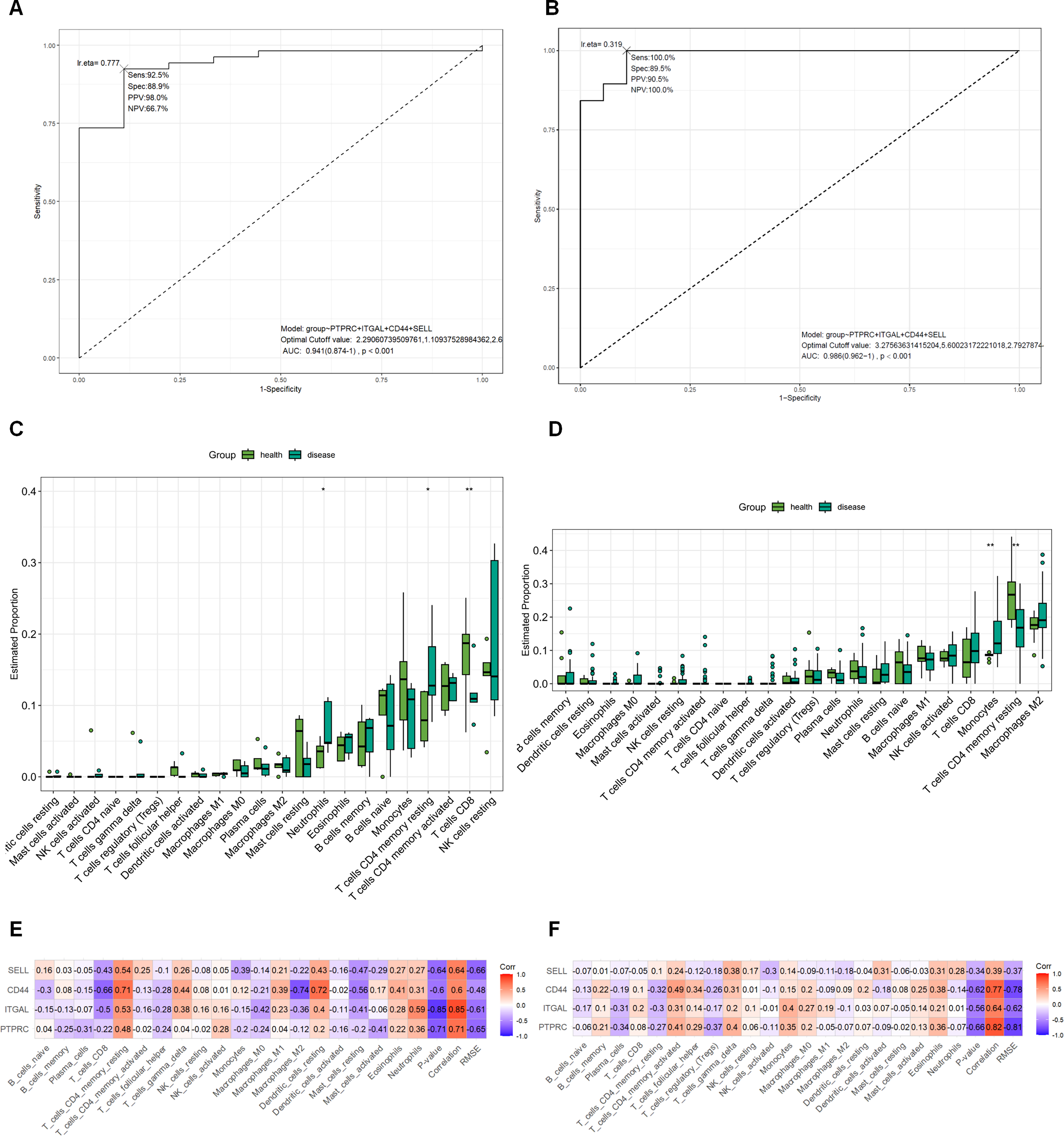
Gut microbiota analysis was performed in fecal samples from NC (n = 10) and SY (n = 10). Chao1 and Shannon indices did not differ significantly between the two groups, and weighted UniFrac PCoA also showed no significant between-group separation (Fig. 3A–C), indicating that global richness, diversity, and overall community structure were not clearly different in this cohort. We therefore interpreted subsequent taxonomic analyses as exploratory assessments of relative compositional differences rather than evidence of broad microbiome restructuring.
Changes in the gut microbiota of AMI patients with CRF. Gut microbiota analysis was performed in fecal samples from the NC group (n = 10) and SY group (n = 10).
Within this exploratory framework, relative abundance analyses suggested differences at the phylum, species, and genus levels between the NC and SY groups (Fig. 3D–F). LEfSe analysis identified 24 differentially represented taxa, with several Enterobacterales-related taxa enriched in the SY group and several taxa including Bifidobacterium-related features enriched in the NC group (Fig. 3G, H). Random forest analysis highlighted Bifidobacterium dentium JCM 1195 DSM 20436 at the species level and Enterococcus at the genus level as potentially discriminative taxa within this small cohort (Fig. 3I, J). However, the corresponding ROC results, particularly the AUC of 1.0 for Enterococcus, should be interpreted cautiously because they were derived from a limited exploratory dataset and may reflect cohort-specific effects rather than reproducible diagnostic performance (Fig. 3K, L). Overall, our microbiome results suggest possible taxa-level compositional differences despite the absence of significant global diversity differences.
Changes in the metabolites of AMI patients with CRF
Untargeted metabolomic profiling was performed in serum samples from NC (n = 10) and SY (n = 10). The integrative microbiota–metabolite correlation analysis was performed in the subset of participants with paired fecal and serum samples (NC, n = 10; SY, n = 10). Metabolic profiling via UHPLC-Q Exactive analysis revealed distinct signatures between the SY and NC groups. PCA demonstrated clear separation along the first two principal components (Fig. 4A), a finding further corroborated by OPLS-DA, which exhibited significant group discrimination (Fig. 4B). Subsequent exploratory screening identified 20 candidate differential metabolites satisfying the predefined inclusion criteria (Fig. 4C, D). Among these, 7 metabolites were significantly upregulated, while 13 were downregulated in the SY group compared with controls. The full list of differential metabolites and their regulation trends is detailed in Supplementary Table S1.
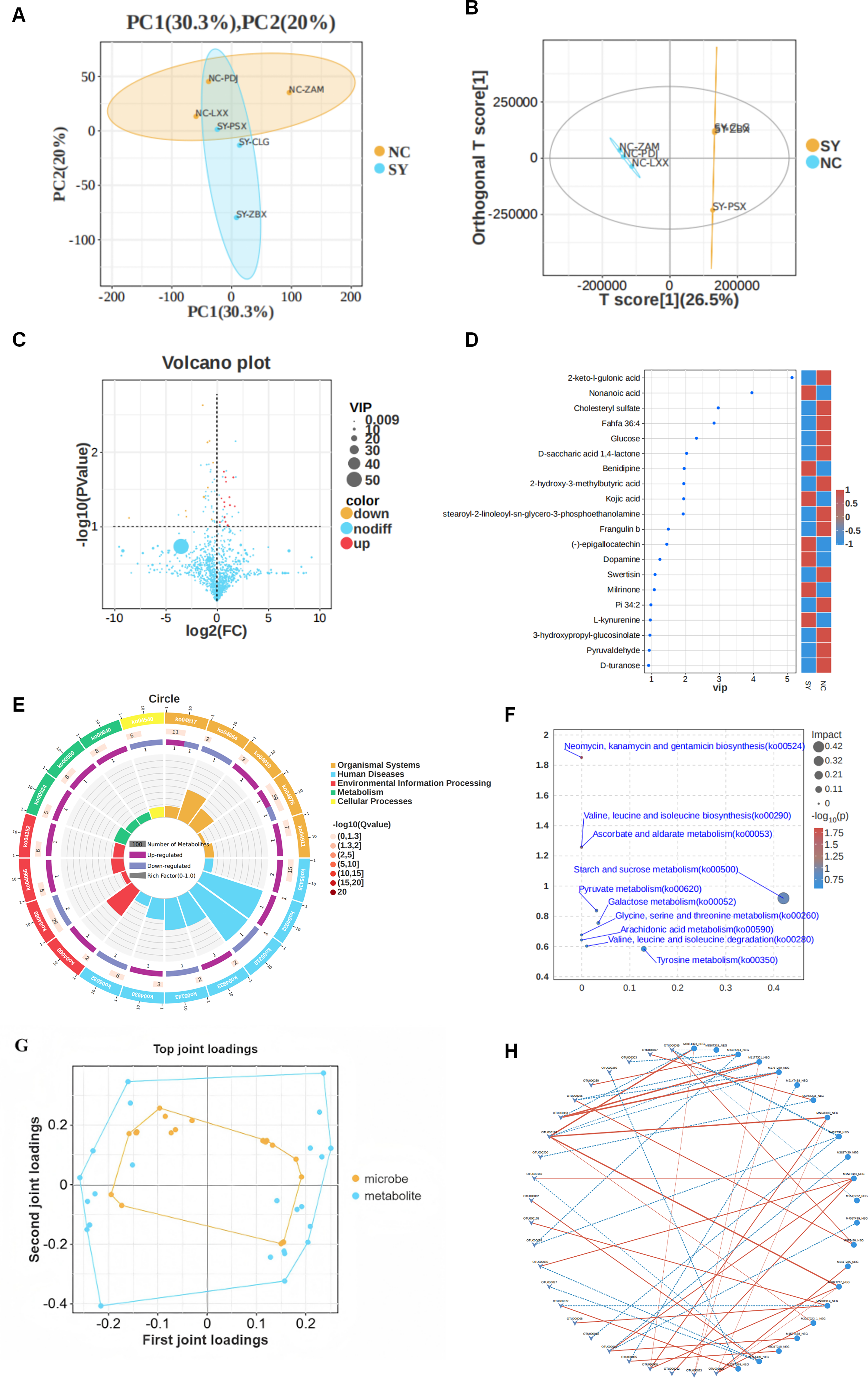
Changes in the metabolites of AMI patients with CRF. Untargeted metabolomic analysis was performed in serum samples from the NC group (n = 10) and SY group (n = 10). The PCA
Functional enrichment analysis (KEGG and MetPA) indicated that these metabolic shifts primarily mapped to amino acid metabolism and carbohydrate/energy metabolism (Fig. 4E, F). To elucidate the functional interplay between the gut microbiome and the metabolome, an O2PLS model was employed to identify features with high joint loadings (Fig. 4G). Correlation analysis further uncovered specific host-microbe interactions (Fig. 4H). Notably, the upregulated metabolite
Discussion
This study used transcriptomics, 16S rRNA sequencing, and untargeted metabolomics to identify candidate multiomic features associated with AMI combined with CRF (Glorieux et al., 2023; Hou et al., 2025; Li et al., 2023). The main findings were as follows. First, four immune-related hub genes, PTPRC, ITGAL, CD44, and SELL, were prioritized from shared AMI and CRF transcriptomic signals. Second, these genes showed preliminary discriminatory performance and were positively correlated with immune cell infiltration. Third, clinical omics analysis suggested selected taxa-level microbiota differences and differential serum metabolites, including increased
PTPRC, also known as CD45, regulates T- and B-cell activation through antigen receptor signaling (Al Barashdi et al., 2021). PTPRC has been identified as an immune-related gene in chronic kidney disease and has been linked to renal injury and immune infiltration (Fang et al., 2024). In AMI, sustained immune activation may promote cytokine release and impair ventricular remodeling (He et al., 2013). In our study, PTPRC showed strong positive correlations with immune infiltration, especially in CRF, suggesting that it may reflect inflammatory activation in the cardiorenal context. ITGAL encodes CD11a, the α-chain of LFA-1, and participates in leukocyte adhesion and migration (Lin et al., 2024). A previous study identified ITGAL as a potential target related to myocardial fibrosis and leukocyte extravasation (Cinato et al., 2025). Our findings suggest that ITGAL may link immune cell trafficking with both myocardial and renal injury. CD44 and SELL are also involved in immune cell migration. CD44 mediates leukocyte adhesion to hyaluronan, whereas SELL regulates T-cell homing to lymph nodes (Zhuo et al., 2006; Yuan et al., 2024). CD44 has also been linked to myocardial ischemia–reperfusion injury and hyperuricemia-induced renal injury through macrophage-related mechanisms (Ma et al., 2024; Zhu et al., 2025). Therefore, co-dysregulation of these genes may indicate a shared leukocyte trafficking program in AMI and CRF. However, these genes remain candidate biomarkers and require experimental and clinical validation.
Immune infiltration analysis supported the relevance of immune dysregulation. In AMI, neutrophils and resting memory CD4 T cells increased, whereas CD8 T cells decreased. Neutrophils are early responders after myocardial infarction and can remove necrotic tissue but may also aggravate tissue injury through reactive oxygen species and protease release (Ma, 2021). CD4 T-cell changes may reflect a pre-existing inflammatory state, while reduced CD8 T cells may indicate impaired immune repair or altered immune balance (Gao et al., 2021; Niederlova et al., 2023). In CRF, increased monocytes and reduced resting memory CD4 T cells were observed. Monocytes can infiltrate renal tissue, differentiate into macrophages, and release profibrotic cytokines, including TGF-β (Tang et al., 2019). Reduced CD4 T-cell function may reflect immune exhaustion in chronic kidney disease (Mathew et al., 2016). The overlap in immune alterations suggests a shared immune vulnerability between the heart and kidney. Renal-derived inflammatory mediators may amplify myocardial injury, while cardiac injury may aggravate renal inflammation (Hundae and McCullough, 2014). Experimental studies also indicate that TLR4/NF-κB/NLRP3 and IDO-related inflammatory signaling may contribute to myocardial or renal injury (Zhuo et al., 2025; Wu et al., 2023). These findings support future mechanistic studies of the hub gene–immune axis.
Microbiome analysis did not show significant alpha- or beta-diversity differences between SY and NC. Therefore, our data do not support broad microbiome restructuring. Instead, they suggest selected taxa-level differences. Enterobacterales-, Enterobacteriaceae-, and Escherichia–Shigella-related signals were relatively enriched in SY, whereas Bifidobacterium-related taxa and Leuconostoc were relatively enriched in NC. This pattern is biologically plausible because Enterobacteriaceae have been associated with uremic toxin generation and systemic inflammation, while Bifidobacterium species are related to short-chain fatty acid production and gut barrier maintenance (Wang and Zhao, 2025; Modrego et al., 2023). Enterococcus may carry inflammatory or virulence-related properties (Daca and Jarzembowski, 2024), but its apparent complete separation in this small cohort should be viewed only as an exploratory signal. Diet, medication use, and recent antibiotic exposure were not adjusted for and may confound microbiome associations.
Metabolomic analysis identified 20 candidate differential serum metabolites.
This study has several limitations. First, the clinical microbiome and metabolomics cohort was small, increasing the risk of false-positive findings and limiting generalizability. Second, the transcriptomic discovery datasets were also limited, especially GSE194388 with only 5 AMI cases and 5 controls. Third, AMI and CRF datasets were analyzed separately rather than as a combined AMI-with-CRF cohort, so shared genes should be interpreted as common immune-related signals rather than disease-specific drivers. Fourth, the ROC results may be optimistic because no prospective cohort, bootstrap validation, or repeated cross-validation was performed. Fifth, the cross-sectional design does not allow causal inference. Sixth, 16S rRNA sequencing provides taxonomic rather than functional information, and microbiome confounders were not adjusted for. Seventh, untargeted metabolomics was not followed by targeted validation, multiple-testing correction was not used in exploratory screening, and detailed QC metrics were incomplete. Finally, animal or cellular validation was not performed. Future studies should include larger independent cohorts, targeted metabolomics, metagenomics, longitudinal outcome data, and experimental validation.
Conclusion
Our multiomics study provides a systematic exploratory analysis of molecular features associated with AMI combined with CRF, identifying candidate hub genes, exploratory microbiota features, and candidate differential metabolites. These findings provide exploratory insights into cardiorenal syndrome and identify candidate biomarkers and candidate pathways for future mechanistic investigation, but the present study does not establish causality or therapeutic utility.
Human Ethics and Consent to Participate Declarations
This study was approved by the Medical Ethics Committee of Hunan Aerospace Hospital (Approval No. HTYY2023LLSH-068-01). All procedures involving human participants were conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all participants prior to enrollment.
Data Availability
The data supporting this study are available from the corresponding author upon reasonable request.
Authors’ Contributions
Huijing L.: Conceptualization, data curation, formal analysis, investigation, methodology, validation, writing—original draft, and writing—review and editing; Huiqiong L., R.W., and M.Z.: data curation, investigation, methodology, and validation.
Footnotes
Author Disclosure Statement
The authors have no conflicts of interest to declare.
Funding Information
The funding needs to revised Sponsored by Science Foundation of Hunan Aerospace Hospital.
Supplemental Material
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.