
Abstract
Malaria caused by Plasmodium falciparum remains a health burden worldwide due to drug resistance and limited treatment options. Calcium-dependent protein kinase 1 (CDPK1) plays a central role in parasite development and invasion, but the downstream molecular alterations that occur upon its disruption remain poorly understood. We present a proteogenomic-based data analysis pipeline for the reanalysis of the publicly available P. falciparum CDPK1 mutant dataset (PRIDE: PXD005207), integrating proteomic and phosphoproteomic data with six-frame genome translation. This led to the discovery of 24 new protein-coding genes, including 17 exonic and 7 intronic peptides, thereby enriching the current genome annotation. Several peptides, such as NILLTFDK, THNNNPQPNPQQK, and EVTSNFGNIR, mapped to previously unannotated genomic regions, which showed orthologous evidence in other Plasmodium species. The reanalysis of phosphoproteomics data identified 37 novel peptides that imply changes in phosphorylation signaling upon CDPK1 knockdown. The identification of conserved peptides like those associated with metacaspase and HSP70, indicates their potential roles in the survival and adaptation of parasites. Overall, this study highlights the potential of proteogenomics to improve genome annotation and reveal hidden coding regions of the P. falciparum genome. This provides new insights into kinase-regulated pathways and potential molecular targets for malaria control.
Introduction
Malaria is a major infectious disease worldwide, caused by Plasmodium parasites, and the most severe and life-threatening cases are caused by Plasmodium falciparum (WHO, 2023). Despite the developments in antimalarial drugs and control of vectors, the disease remains responsible for high morbidity and mortality, particularly in tropical regions. The increase in drug-resistant parasites and insecticide-resistant mosquitoes also complicates eradication (Ashley et al., 2014). A better understanding of the molecular processes that regulate parasite development, differentiation, and host invasion is essential for identifying novel therapeutic approaches.
Protein kinases are key regulators of P. falciparum biology, mediating processes such as cell cycle progression, invasion, egress, and sexual differentiation (Doerig et al., 2015; Tewari et al., 2010). Among them, CDPK1 is one of the most well-characterized kinases of P. falciparum. CDPK1 acts as a primary regulator of calcium-dependent signaling, coordinating processes essential for parasite motility and invasion of host erythrocytes (Kumar et al., 2019). It phosphorylates several substrates of the glideosome complex, a motor apparatus required for host cell invasion. It cooperates with cyclic AMP-dependent protein kinase (PKA) to control intracellular signaling pathways (Bansal et al., 2013). Disruption of CDPK1 impairs parasite invasion and growth, emphasizing its pivotal role in P. falciparum pathogenesis (Green et al., 2008). However, the downstream molecular and compensatory effects of CDPK1 disruption remain incompletely elucidated. Mass spectrometry (MS)-based proteomics has improved the knowledge of P. falciparum proteome, post-translational modifications, and signaling (Lasonder et al., 2012; Pease et al., 2013). The genome of P. falciparum exhibits an exceptionally high AT content based on the plasmoDB release 67 assemblies, which is consistent with the dataset employed in the present study (Gardner et al., 2002; Otto et al., 2014). Conventional proteomics tends to miss peptides from unannotated regions. Proteogenomics, which integrates proteomic, genomic, and transcriptomic information, helps improving genome annotations and discovering new protein-coding regions (Menschaert and Fenyö, 2017; Nesvizhskii, 2014). Proteogenomic analysis identifies genome search-specific peptides (GSSPs) that map to unannotated regions of the genome, describing new exons, intronic peptides, or extended coding sequences (Datta et al., 2016; Prasad et al., 2017). In P. falciparum, the six-frame translation of the genome, combined with high-resolution MS/MS spectra, has revealed new peptides and refined gene models, validating translation in areas earlier defined as noncoding (Sims and Hyde, 2006).
The publicly deposited P. falciparum CDPK1 mutant dataset (Kumar et al., 2017) comprises iTRAQ-labeled proteomic and phosphoproteomic data originally employed to investigate the molecular mechanism mediated by CDPK1. In the current work, the data were reanalyzed using a proteogenomic pipeline that searched proteomic and phosphoproteomic data with a six-frame translated genome to identify previously unreported peptides and coding regions. Such analysis has the potential to reveal compensatory or cryptic gene products arising due to CDPK1 disruption, shedding light on adaptive regulatory strategies and evolutionary conservation across Plasmodium species.
This research underscores the use of proteogenomics to refine the genome annotation and reveal the hidden coding potential of genomic regions in P. falciparum. Discovery of new peptides, exonic extensions, and intronic regions sheds light on the parasite’s translational landscape and adaptive strategies for kinase perturbation. Here, we reanalyzed the raw data using the current Plasmodium falciparum 3D7 (version 67) reference, with a specific focus on identifying CDPK1 kinase-related proteins that may serve as anti-malarial targets. Overall, these results contribute to a more integrated understanding of kinase-dependent signaling and will potentially aid in the identification of new molecular targets for malaria intervention (Antil et al., 2021; Arefian et al., 2022; Prasad et al., 2012).
Methods
Dataset and database search
The MS data were obtained from the Proteomics Identification Database (PRIDE) repository. The raw data were accessed at the PRIDE identifier: PXD005207 (Kumar et al., 2017), which contains the iTRAQ-labeled proteomic and phosphoproteomic datasets. The raw data consisted of six datasets (proteomic and phosphoproteomic datasets) with a total of 120 raw files. All raw files were analyzed using Proteome Discoverer (PD) version 2.2 and searched with SequestHT and Mascot algorithms. Initial search was performed against the P. falciparum reference proteome database of the PlasmoDB Release 67 (5386 protein sequences), Homo sapiens protein database (National Center for Biotechnology Information [NCBI] RefSeq Version 110) and a common Repository of Adventitious Proteins (cRAP) database (116 contaminant protein sequences). In addition, an in-house Python script (Narayana, 2024) was used to generate a six-frame translation of the P. falciparum genome.
Carbamidomethylation of cysteine (C) residue was specified as a fixed modification, along with iTRAQ labeling at the peptide N-terminus and lysine (K) residue. Methionine (M) oxidation and N-terminal protein acetylation were specified as dynamic modifications. For phosphoproteomic datasets, phosphorylation (+79.99 Da) at serine (S), threonine (T), and tyrosine (Y) was also included as a variable modification. Trypsin was selected as a protease digestion enzyme, with a maximum of two missed cleavages. A precursor mass tolerance of 20 ppm and a fragment mass tolerance of 0.1 Da were applied. Peptide-spectrum matches (PSMs) were filtered at a false discovery rate (FDR) of 1%, and the percolator algorithm was employed to calculate the posterior error probability for each PSM, thereby assigning statistical confidence to each spectral match.
Proteogenomic analysis workflow
A six-frame translation of the P. falciparum genome was carried out to generate sequences between stop-stop codons with a minimum length of seven amino acids. This is based on commonly accepted proteomics search parameters and previously published proteogenomic studies (Rex et al., 2022). The unassigned MS/MS spectra were searched against the custom databases using the same criteria utilized in proteome analysis. The novel peptides identified in proteogenomic analysis were confirmed using the Basic Local Alignment Search Tool (BLASTp) (Datta et al., 2016). Protein–protein BLAST search against PlasmoDB and NCBI databases (Altschul et al., 1990; Aurrecoechea et al., 2009). Novel proteins of the orthologous regions in related species were further confirmed using ClustalW (Chenna et al., 2003).
Using proprietary Python programs, GSSPs were categorized as novel protein-coding genes and gene corrections. The peptides that met the 1% FDR criterion and showed unique genome matches were included. In order to determine the genomic location of the sequences (exonic, intronic, or 5′-extension), the P. falciparum genome was mapped with the integrated genome viewer (IGV), which allowed the mapping of identified peptides and transcript regions (Robinson et al., 2011). Figure 1 depicts the complete proteogenomic workflow, including data acquisition from PRIDE, database searching with PD 2.2, extraction of unassigned spectra, six-frame genome translation, and unique peptide validation by BLAST.
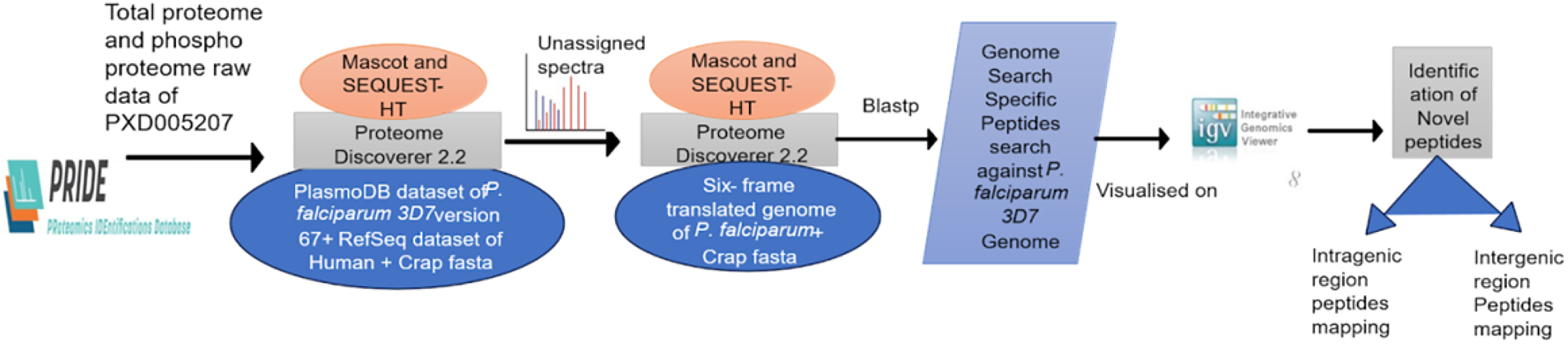
Workflow of the study: CDPK1 mutant proteomics and phosphoproteomics raw data taken from PRIDE were initially searched against the protein databases in Proteome Discoverer 2.2 using Mascot and SEQUEST-HT. The unassigned spectra were then searched against a six-frame translated genome database, unique peptides were identified using protein BLAST, and the results were visualized in the Integrated Genome Viewer (IGV).
Results
Proteomic and phosphoproteomic identification from the protein database and six-frame translation
Proteogenomic analysis of MS data identified 24 new protein-coding genes, 17 novel exons, and 7 intronic sequences. The datasets used in this study were previously focused on phosphorylation modifications and identified key phosphosites. However, in the current study, we explored it to identify novel peptides by searching against the six-frame translated genome database. A total of 1,084,084 MS/MS spectra were searched against the proteome database, yielding 252,996 PSMs and 28,029 peptides corresponding to 2865 proteins. The remaining 911,977 MS/MS unassigned spectra were searched against the six-frame translated genome database, and 864,793 spectra were assigned to 4,458 PSMs, corresponding to 1,965 peptides and 831 proteins. BLASTp analysis of these peptides uncovered 16 peptides that failed to match any previously identified Plasmodium falciparum 3D7. The compilation and genomic distribution of the new peptides are shown in Table 1 and Figure 2, respectively.
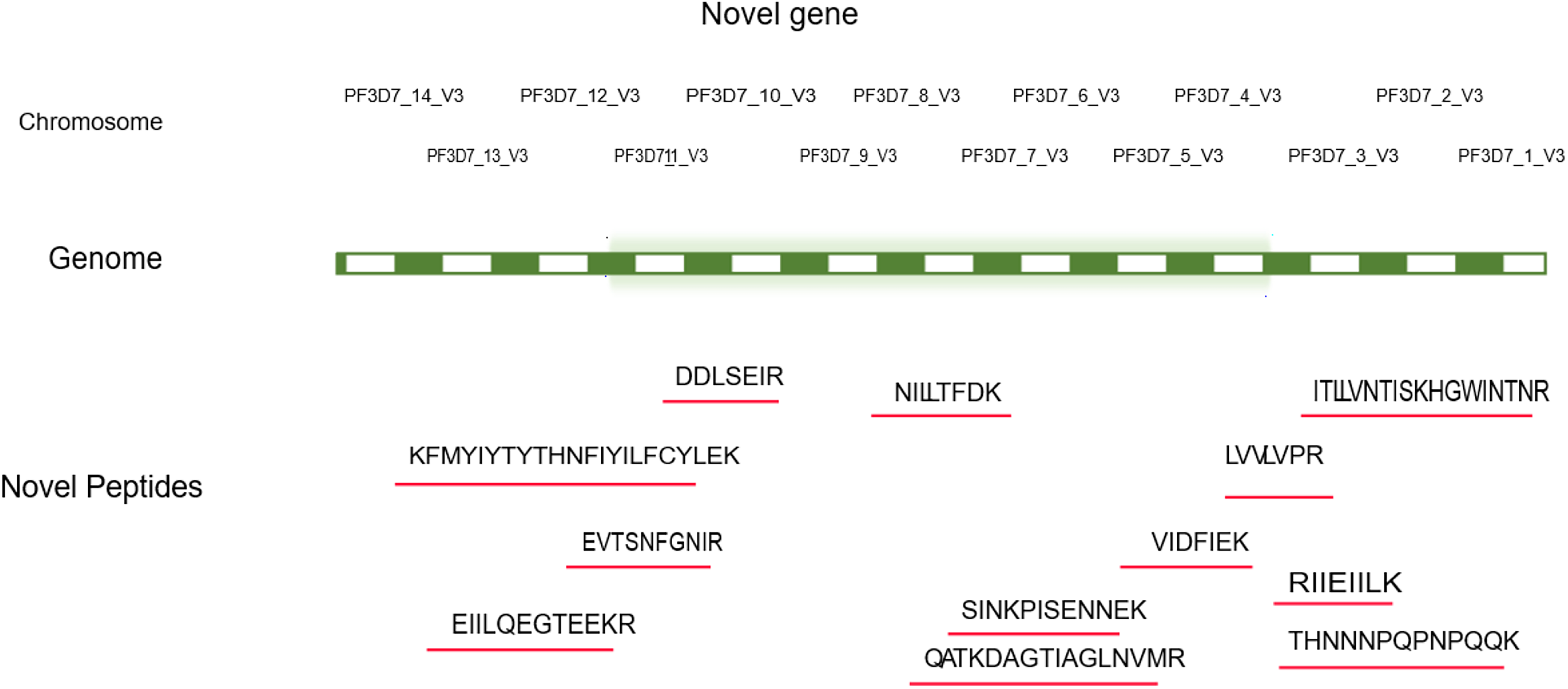
Peptides mapping to the P. falciparum genome was performed using IGV to identify novel protein-coding genes.
Partial list of novel peptides identified in the CDPK1 mutant proteomics Dataset

In the phosphoproteomic search, 8,02,677 MS/MS spectra were identified and assigned to 13,904 PSMs and 890 peptides corresponding to 877 proteins. Searching the remaining 7,69,351 MS/MS spectra against the six-frame translated genome database generated assigned to 229 PSMs with 83 peptides corresponding to 141 proteins. BLASTp analysis of this phosphor dataset identified 37 novel peptides.
Visualization of GSSPs in IGV
The peptides were mapped to the P. falciparum genome and visualized using IGV with the respective transcript GTF file to determine whether they originated from exonic, intronic, or any other regions. Of the 24-GSSPs, 17 mapped to the exonic region and the remaining 7 originated from intronic sequences. Additional information is mentioned in Supplementary Table S1. The novel peptide NILLTFDK, which is translated from the exonic region of chromosome 8, corresponds to a 30-amino-acid protein encoded by the gene PF3D7_0819600 (putative thionin 2.4). The MS/MS spectrum demonstrated well-resolved y- and b-ion series at m/z value of 626.38, confirming its peptide identity (Fig. 3A and B). Other novel peptide spectra are submitted in Supplementary Figure S1.
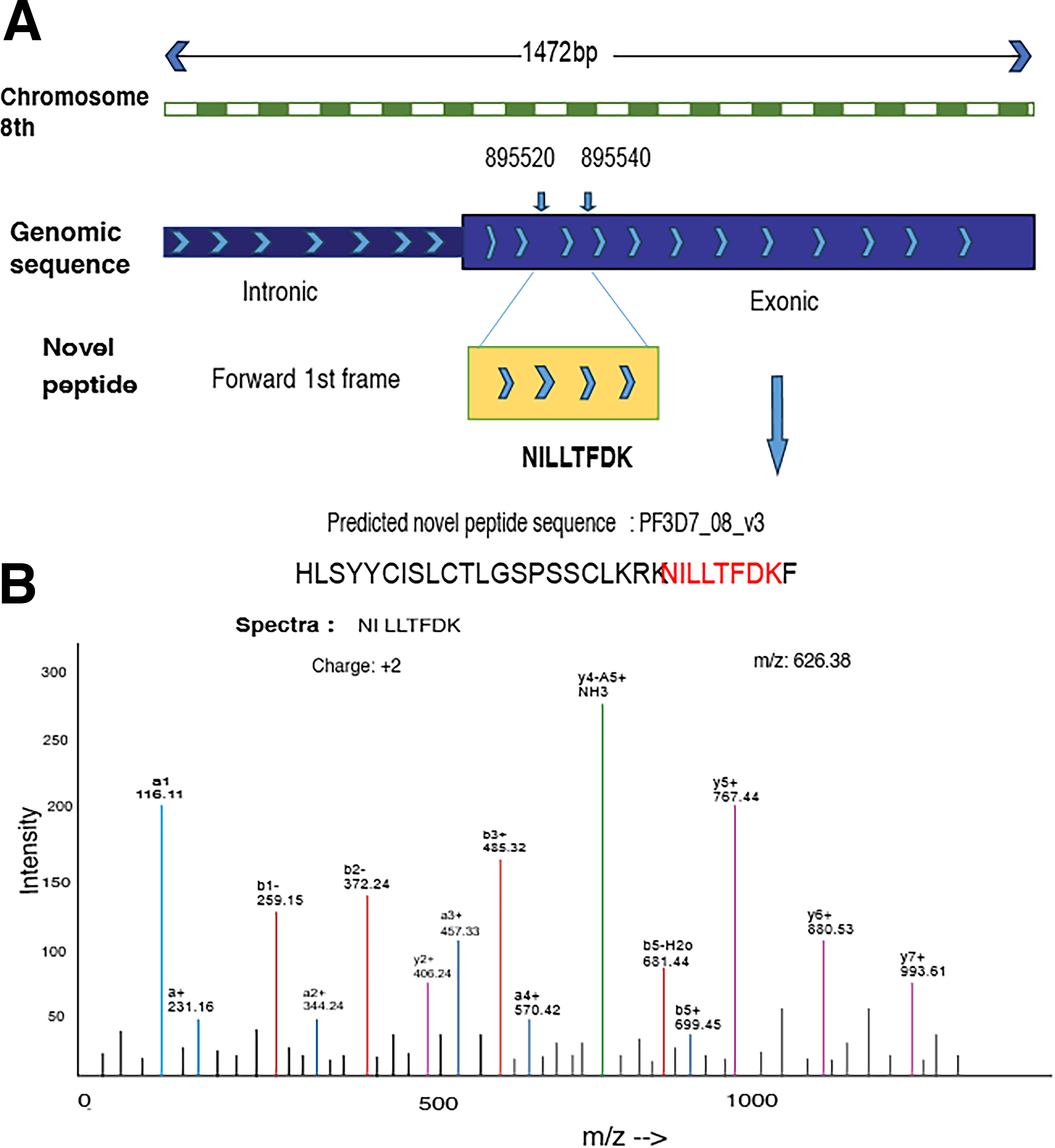
Orthologous evidence and C-terminal extension of the novel gene
Using ClustalW, we identified multiple peptides that exhibit orthologous evidence with proteins from other Plasmodium species and even higher eukaryotes. For example, the peptide QATKDAGTIAGLNVMR showed orthologous evidence in P. malariae, P. vivax, and P. berghei. This peptide corresponds to the HSP70 protein, which has also shown evidence in Arabidopsis thaliana and Theileria parva (Supplementary Fig. S2A). Orthologous evidence is given in Supplementary Table S1. In addition, four GSSPs were identified in the exonic region between the genes Pf3D7_05001001 and PF3D7_05321001 on chromosome 5 of P. falciparum 3D7. Out of them, three peptides (IDLSIASINELSK, LLLLPR, and THNNNPQPNPQQK) are C-terminal exon extensions (GSSP13, GSSP16, and GSSP8), indicating additional amino acids at the C-terminal end of known protein termini (Supplementary Fig. S2B).
GSSPs identification in the P. falciparum genome
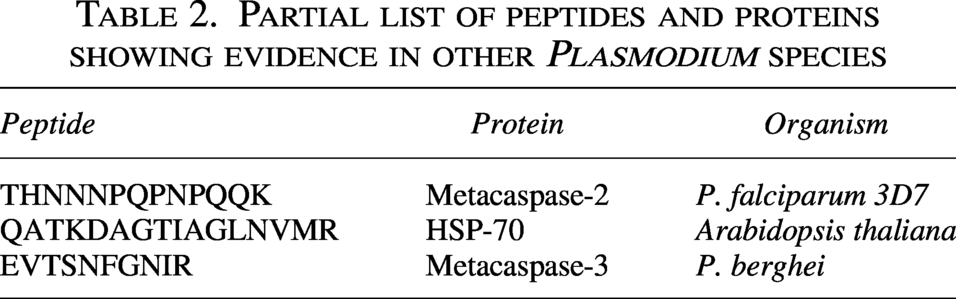
We found evidence for 16 GSSPs in the P. falciparum genome. Of these, 11 peptides were novel for P. falciparum 3D7 species, whereas 5 peptides were conserved across other Plasmodium species. Interestingly, the peptide EVTSNFGNIR corresponds to Metacaspase-3. Table 2 shows a partial list of peptides and proteins that are present in other Plasmodium species.
Partial list of peptides and proteins showing evidence in other Plasmodium species
Discussion
CDPK1 plays a major role in developmental regulation and host cell invasion of P. falciparum. CDPK1 phosphorylates a number of subunits of the glideosome complex and also interacts with PKA to coordinate parasite motility and erythrocyte invasion (Kumar et al., 2017). Perturbation of CDPK1 function can thus have downstream signaling consequences and transcriptional regulation.
In the present study, we performed a systematic proteogenomic analysis of P. falciparum 3D7, with a particular focus on the CDPK1 mutant dataset. By integrating both proteomic and phosphoproteomic data with six-frame genome translation, we identified several novel peptides and potential coding regions that were previously unannotated in the P. falciparum genome. It includes new protein-coding regions, exonic extensions, and intronic peptides. These findings demonstrate the ability of proteogenomic methods to improve genome annotation of P. falciparum and to identify hidden genomic elements that may be responsible for the parasite’s adaptive biology.
In this study, proteomic analysis detected more than 28,000 peptides for 2865 proteins, accounting for about 54% of the P. falciparum proteome. Significantly, six-frame translation-based searches found 1965 peptides for 831 proteins, including 24 putative novel protein-coding genes, 17 novel exons, and 7 intronic sequences. These results demonstrate the capability of proteogenomics in discovering hidden regions of coding potential beyond conventional annotation.
The identification of intron-derived and exon-extended peptides such as NILLTFDK (from chromosome 8, mapped to PF3D7_0819600) validates the hypothesis that uncharacterized transcriptional and translational events are likely to play regulatory functions in parasite gene regulation. Detection of C-terminal exon extensions in peptides such as THNNNPQPNPQQK and LLLLPR also indicates structural diversification among protein isoforms, potentially contributing to functional adaptation and stage-specific expression.
Among the identified novel peptides, the peptide THNNNPQPNPQQK, homologous to metacaspase-2 (PF3D7_1438400), is particularly interesting because it plays a role in gametocyte development and parasite transmission. Previous studies in P. berghei have shown that targeting metacaspase-2 results in decreased gametocyte production as well as ineffective transmission (Kumari et al., 2022). Loss of metacaspase-2 disrupts gametogenesis, leading to reduced gametocyte development, fewer oocysts and sporozoites, and delayed liver-stage invasion. It is identified here that P. falciparum supports the conserved function of meta caspases in the regulation of parasite life cycles (Vandana et al., 2018,2020). Similarly, the peptide QATKDAGTIAGLNVMR, which aligns with HSP70 proteins and shows orthology with P. vivax, P. malariae, and P. berghei, indicates conservation of molecular chaperone mechanisms in Plasmodium species. HSP70 proteins have been implicated in protein folding, stress response, and drug resistance, which may be significant for parasite survival under host immune stress or exposure to antimalarial drugs (Shonhai, 2021).
The phosphoproteomic data identified 37 new peptides, some of which were present only in wild-type but not in the CDPK1 mutant. The differential peptide profile indicates the potential regulation of phosphorylation-dependent signaling pathways by CDPK1. For example, peptides such as YIIKIDTMFQISSRDIQLCSCFVSSHLWDSYPL and FSNLYLFSSFFFFLSSSSSLFK were found exclusively in control samples, suggesting their downregulation following CDPK1 disruption. Such variations may reflect CDPK1-mediated phosphorylation reactions or indirect regulatory mechanisms affecting protein stability or expression.
The detection of peptides from intronic regions such as EVTSNFGNIR corresponds to metacaspase-3, suggesting functional overlap between proteolytic cascades involved in parasite differentiation and stress response and metacaspase-3 (PF3D7_1416200) was serine-like protease predominantly expressed in the sexual stage (Kumar et al., 2019; Kumari et al., 2022). The identification of new peptides within the CDPK1 mutant background could be a potential stage-specific biomarker of parasite activity or drug resistance. In addition, understanding how CDPK1 disruption modulates other signaling pathways could reveal new therapeutic targets within the parasite kinome and phosphor regulation network. However, in the present study, functional annotation for certain GSSPs was inferred based on the sequence similarity and database annotations. Additional experimental validation and orthogonal supporting data are necessary for conclusive functional identification, as some short peptides may be conserved across different proteins or Plasmodium species.
Although this study offers new insights into the P. falciparum gene annotation under CDPK1 knockdown through proteogenomic analysis, it has certain limitations. The mass spectrometer may miss low-abundance or hydrophobic peptides, and the function of the newly discovered peptides also requires experimental verification. In the current study, proteogenomic analysis was performed using PlasmoDB Release 67 and solely focused on the previously generated iTRAQ data (PXD005207) digested with trypsin. As PlasmoDB is updated regularly, some of the newly discovered genomic locations reported in this work may be included in future versions of the database. In addition, the use of tryptic peptide-based proteogenomics may prevent the detection of hidden coding regions that lack Lysine/Arginine cleavage sites. Future studies involving proteolysis with additional proteases would enable even better detection of previously uncharacterized protein-coding regions in Plasmodium falciparum. Structural modeling and functional domain prediction would provide further insight into the novel protein-coding genes identified. However, functional annotation including domain prediction and structural modeling will be considered in our future investigation. Applying the same approach to other life stages or drug-resistant parasites could give further insight into parasite adaptation and evolution.
Conclusions
In summary, the present study on the P. falciparum CDPK1 mutant revealed the evidence of novel protein-coding genes, new exons, and intronic peptides. These discoveries improve the annotation of the P. falciparum genome and highlight the complexity of its post-transcriptional and post-translational regulation systems. The presence of conserved and functionally significant peptides, such as those linked to metacaspase and HSP70, emphasizes their potential roles in survival, transmission, and pathogenesis of the parasite. Overall, the current study demonstrates how proteogenomics can be used to reveal unannotated gene products, uncover the malaria parasite’s latent coding potential and advance our understanding of its molecular biology.
Authors’ Contributions
N.C. performed the analysis and article drafting. A.B.R performed the analysis. T.S.K.P. and S.D. conceptualized the study, designed the components, and reviewed/corrected the article. All authors read and approved the final version of the article.
Footnotes
Acknowledgments
The authors acknowledge the Department of Biotechnology (DBT) National Facility grant for supporting the Center for Systems Biology and Molecular Medicine, mainly through the project “Skill Development in Mass Spectrometry-based Metabolomics Technology BIC” (BT/PR40202/BTIS/137/53/2023). The authors also thank the ICMR for designating our Center as the ICMR-Collaborating Center of Excellence 2024 (ICMR-CCoE) in recognition of the commendable achievements in biomedical research.
Author Disclosure Statement
The authors declare no conflict of interest.
Funding Information
No funding was received for this article.
Supplemental Material
Supplemental Material
Supplemental Material
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.