
Abstract
Geroscience offers a transformative paradigm by targeting shared aging hallmarks to enable simultaneous modulation of multiple age-related disorders (ARDs). Yet, current geroprotective interventions often lack mechanistic breadth, as targeting isolated pathways yields limited benefits compared to interventions modulating interconnected regulators of aging biology. To bridge this gap, a systems-level strategy was designed around four key targets, including, Nrf2/Keap1, mTORC1, AMPK, and SIRT1, responsible for regulating oxidative stress, mitochondrial dysfunction, proteostasis, and autophagy. Concurrent regulation of these targets was identified to potentially induce a concerted and sustained geroprotective effect across diverse ARDs. A machine learning-based geroprotector classification model was developed to identify natural compounds capable of executing this integrated strategy. Subsequent drug-likeness screening confirmed favorable pharmacokinetic properties of the predicted compounds, while molecular docking revealed compounds with strong binding affinities with all four geroprotective targets, thereby leading to the identification of a subset of natural compounds with the potential to induce a coordinated geroprotective response. Finally, a graph neural network-based synergy prediction model, trained on known ARD drug combinations, identified five high-confidence pairs composed of four natural compounds, including Baicalein, Pectolinarigenin, Phloretin, and Demethoxycurcumin. These computationally predicted combinations hold the potential to elicit synergistic and comprehensive geroprotective effects across multiple ARDs.
Introduction
The rising incidence rate of age-related disorders (ARDs) including neurological disorders, type 2 diabetes, heart diseases and chronic kidney disease due to global population aging has driven significant interest in geroscience and the development of geroprotective therapeutic strategies. Geroprotective approaches, which aim to delay the onset or progression of ARDs, offer a comprehensive solution by addressing the underlying biology of aging rather than merely managing disease symptoms (Fekete et al., 2024; Kroemer et al., 2025). By targeting the intricate network of aging-related pathways, these strategies enable the restoration and maintenance of cellular homeostasis, making these strategies concurrently applicable in the treatment of all ARDs. However, most of these geroprotective therapeutic strategies tend to address individual aspects of the aging biology, which has highly complex and intricate nature and thus the effectiveness and sustainability of such geroscience-based strategies fail to compete with that of standard therapeutic interventions (Cummings et al., 2023; Forman et al., 2023; Kroemer et al., 2025; Rolland et al., 2023). Considering the highly dynamic and interconnected nature of processes and pathways involved in the intricate network of the biology of aging and its association with several diseases, it is necessary to devise such geroprotective interventions capable enough to address each hub of this interconnected network and thereby induce a comprehensive and sustainable therapeutic effect in multiple ARDs. In order to devise such all-encompassing strategies, extensive literature review was conducted to identify the primary hubs of the aging biology network involved in ARD progression as well as in the associated therapeutics and it was observed that processes including oxidative stress, mitochondrial dysfunction, proteostasis (including proper protein folding and efficient protein degradation) and autophagy, play a crucial role in the onset and progression of ARDs and have been explored for their individual therapeutic potential multiple times (Cheon et al., 2019; Hipp et al., 2019; Kumar et al., 2023; Santos et al., 2024; Somasundaram et al., 2024; Tabibzadeh, 2023; Wu et al., 2024).
Mitochondrial dysfunction, oxidative stress, impaired proteostasis and defective autophagy are highly interconnected hallmarks of aging that collectively contribute to the onset and progression of ARDs. Aging-associated mitochondrial dysfunction increases reactive oxygen species (ROS) production, causing oxidative damage that further exacerbates mitochondrial impairment and disrupts proteostasis (Somasundaram et al., 2024). Simultaneously, declining efficiency of molecular chaperones, the ubiquitin-proteasome system and autophagy promotes accumulation of damaged proteins and organelles, thereby aggravating cellular dysfunction and ARD progression (Kumar et al., 2023; Tabibzadeh, 2023). Since autophagy is responsible for clearance of dysfunctional proteins and mitochondria, its decline further amplifies oxidative stress, mitochondrial dysfunction and proteostatic imbalance through interconnected feedback mechanisms (Aran and Singh, 2023; Lévy et al., 2019; Ornatowski et al., 2020).
Given these cyclical and intricately cross-regulated sub-networks, their deeper mechanistic exploration led to the identification of four key components: Nuclear factor erythroid 2-related factor 2 (Nrf2)/Kelch-like ECH-associated protein 1 (Keap1), mammalian target of rapamycin complex 1 (mTORC1), Sirtuin 1 (SIRT1) and AMP-activated protein kinase (AMPK). Nrf2, a master regulator of antioxidant response, is normally sequestered in the cytoplasm by Keap1, leading to its ubiquitination and proteasomal degradation (Ngo and Duennwald, 2022). Under oxidative stress, Nrf2 dissociates from Keap1, translocates to the nucleus and activates transcription of antioxidant enzymes and proteasomal components, regulating both redox balance and proteostasis (Monsalvo-Maraver et al., 2024). Conversely, mTORC1 hyperactivation suppresses autophagy, causing buildup of dysfunctional mitochondria and protein aggregates (Guillén and Benito, 2018; Querfurth and Lee, 2021); its inhibition, for instance by rapamycin, has extended lifespan in model organisms and remains a key geroprotective target (Konopka et al., 2023; Mannick and Lamming, 2023). In contrast, SIRT1 and AMPK regulate oxidative stress, mitochondrial homeostasis, proteostasis, and autophagy through multiple interconnected pathways and also form a positive feedback loop (Abu-Baih et al., 2024; Marino et al., 2021; Ottens et al., 2021; Ruderman et al., 2010; Sacitharan et al., 2020; Singh et al., 2018; Wang et al., 2022; You and Liang, 2023). Together with Nrf2 activation and mTORC1 inhibition, these regulators collectively coordinate antioxidant defense, autophagy, mitochondrial quality control and proteostasis through interconnected mechanisms involving lysosomal biogenesis, mitophagy, proteasomal regulation and cellular stress-response pathways (Armeli et al., 2024; Białopiotrowicz-Data et al., 2023; Esteras and Abramov, 2022; Garcia and Shaw, 2017; Herzig and Shaw, 2018; Huang et al., 2024; Laplante and Sabatini, 2012; Lee, 2019; Monsalvo-Maraver et al., 2024; Ngo and Duennwald, 2022; Panwar et al., 2023). While each of these regulators can independently modulate aging-associated cellular processes, their mechanistic overlap and coordinated regulation suggest that simultaneous activation of Nrf2, SIRT1, and AMPK alongside inhibition of mTORC1 could potentially induce synergistic restoration of cellular homeostasis across multiple ARDs through integrated modulation of interconnected aging-associated pathways (Fig. 1).
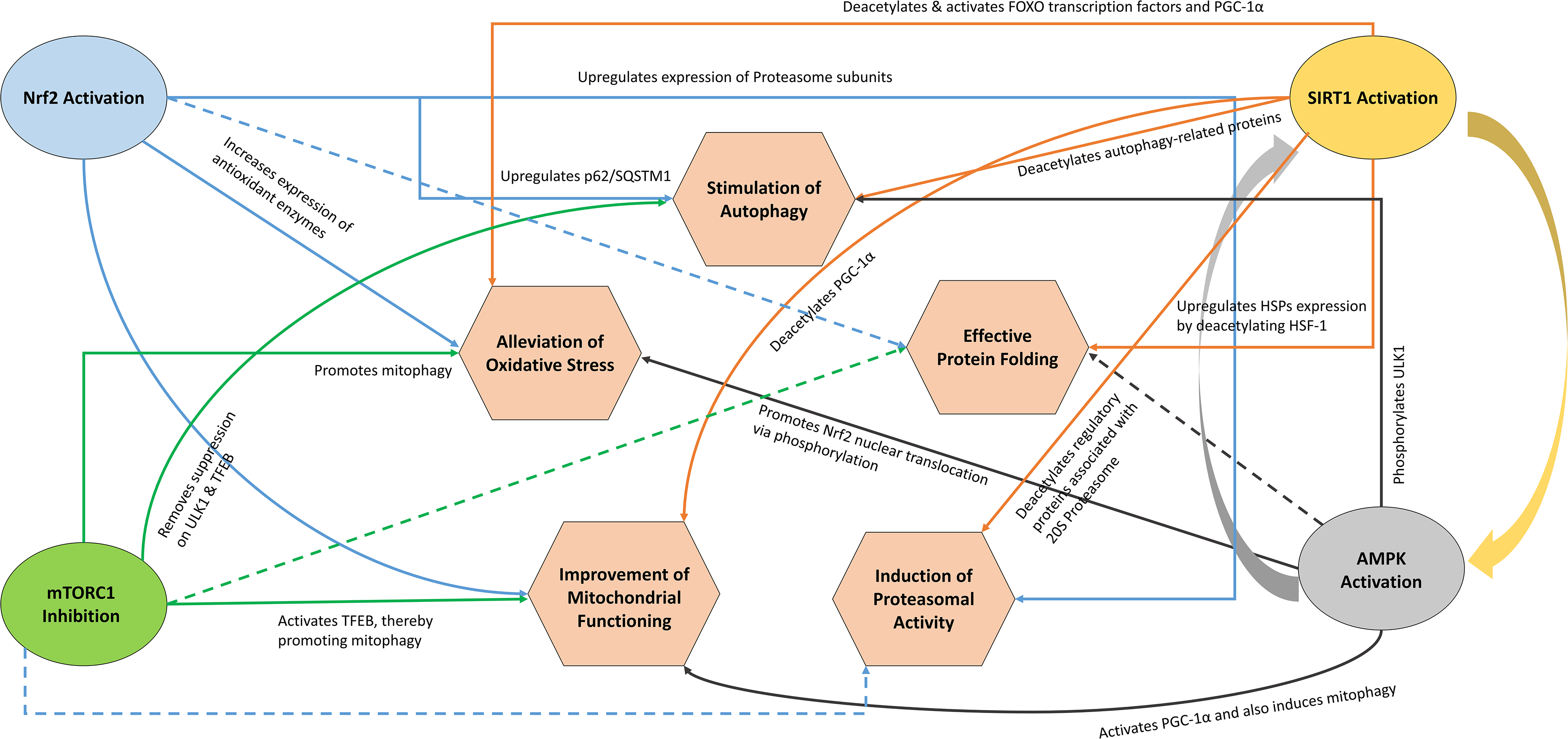
Synergistic interplay associated with concurrent Nrf2 activation, mTORC1 inhibition and SIRT1 and AMPK activation. The solid lines represent a direct role in the regulation of a particular process, whereas dashed lines represent indirect regulation. The curved arrows between SIRT1 activation and AMPK activation represent a positive feedback loop. AMPK, AMP-activated protein kinase; SIRT1, sirtuin 1.
This cooperative engagement of geroprotective pathways forms a strong conceptual foundation for combinatorial therapeutic strategies. Targeting these four regulators in unison offers a promising approach to mitigate age-associated cellular dysfunction, thereby restoring cellular homeostasis and ultimately inducing comprehensive therapeutic effects in multiple ARDs. And in line with this therapeutic strategy, the present study introduces a computational framework developed to screen potential geroprotective combinations of natural compounds (phytochemicals) with the ability to simultaneously target all the aforementioned factors including Nrf2, mTORC1, SIRT1, and AMPK. Here, machine learning-based geroprotector classification and molecular docking were conducted to identify natural compounds with geroprotective characteristics and computationally predicted ability to concurrently engage with all the desired targets in order to modulate their intricate network. Subsequently, a graph neural network (GNN)-based synergy prediction model was developed to predict potential combinations among the screened natural compounds.
Materials and Methods
Dataset preparation, data preprocessing, and machine learning-based classification
A dataset comprising 466 FDA-approved drugs was constructed using the DrugBank database and thorough literature review (Knox et al., 2024). The dataset included drugs approved for treating ARDs, such as neurological disorders, type 2 diabetes, heart diseases and chronic kidney disease, as well as non-ARDs (NARDs), including infectious diseases, cancer and various genetic disorders. The drugs were assigned numerical codes based on their application in different diseases, resulting in a dataset with 466 FDA-approved drugs labeled with numerical values including “0” for NARDs and 1’ for ARDs (Supplementary Table S1). Thereafter, 1024-bit molecular fingerprints were calculated for all the drugs using RDKit and these fingerprints were then used as features to train the classification model (Landrum and Greg, 2006). Fingerprints were standardized by z-score normalization and dimensionality reduction was performed using Principal Component Analysis (PCA), retaining 50 components that captured the majority of dataset variance while minimizing noise and improving computational efficiency. The dataset was split into training and testing sets in an 80:20 ratio and multiple classifiers, including Random Forest, Decision Tree, Support Vector Machine (SVM), Logistic Regression, k-Nearest Neighbors (KNN), Gradient Boosting, and XGBoost, were trained and evaluated (Chen and Guestrin, 2016; Pedregosa et al., 2011). Model performance was assessed through classification metrics and receiver operating characteristic (ROC) analysis, complemented by five-fold stratified cross-validation for robustness evaluation. In addition, external validation was performed using a dataset of 144 geroprotectors retrieved from the Geroprotectors.org database, which contains compounds with experimentally reported lifespan-extending and geroprotective effects across various ARDs (Moskalev et al., 2015). Only those compounds were selected that were not present in the training or test datasets to ensure an unbiased evaluation. Molecular fingerprints were generated for all compounds, followed by the same preprocessing pipeline used during model development, including normalization and PCA. The trained model was then applied to this dataset and its performance was evaluated in terms of prediction accuracy, representing the coverage of known geroprotectors.
Chemical space projection of FDA-approved ARD drugs and predicted geroprotective natural compounds
In order to assess potential distribution shift between the FDA-approved ARD drugs used for training the classification model and natural compounds predicted as geroprotectors and to further validate the robustness of the geroprotector classification model, chemical space projection was performed using Uniform Manifold Approximation and Projection (UMAP) (McInnes and Healy, 2018). Molecular fingerprints were used as representations/features for each compound and were then projected into a two-dimensional plot using UMAP to visualize their relative distribution and structural relationships. This analysis was used to examine the overlap, proximity, and potential extensions between the two groups, thus generating the qualitative assessment of model generalizability across structurally diverse compounds.
Screening of natural compounds for drug-likeness
After screening the natural compounds for their geroprotective roles using the classification model, the natural compounds predicted as geroprotectors were further evaluated for drug-likeness using SWISS ADME (Daina et al., 2017). Compounds were analyzed for their adherence to Lipinski’s Rule of Five and those with any violations were excluded (Lipinski et al., 2001).
Molecular docking of natural compounds with geroprotective targets
The 3D structures of the target proteins were retrieved from the Protein Data Bank (PDB) (Berman et al., 2000), with PDB IDs as follows: Keap1 (PDB ID: 6QME), mTORC1 (PDB ID: 8ERA), AMPK (PDB ID: 5KQ5) and SIRT1 (PDB ID: 4ZZH). Protein preparation was performed in PyMOL (Schrödinger LLC, 2015), involving the removal of water molecules, bound ligands, and unnecessary chains, along with the addition of polar hydrogens.
The 3D conformers of the natural compounds were obtained from PubChem (Kim et al., 2025), and docking was conducted using the AutoDock Vina program (Trott and Olson, 2010) within the PyRx GUI (Dallakyan and Olson, 2015). The grid box parameters for each protein were as follows:
Binding affinities (in kcal/mol) were calculated for the natural compounds across the four proteins, providing critical insights into their potential as geroprotectors. Top candidates were then identified by applying a binding affinity filter. The selected candidates were subsequently subjected to site-specific redocking validation using BIOVIA Discovery Studio (DASSAULT SYSTÈMES, 2025).
Drug pair dataset preparation for GNN-based synergy prediction model
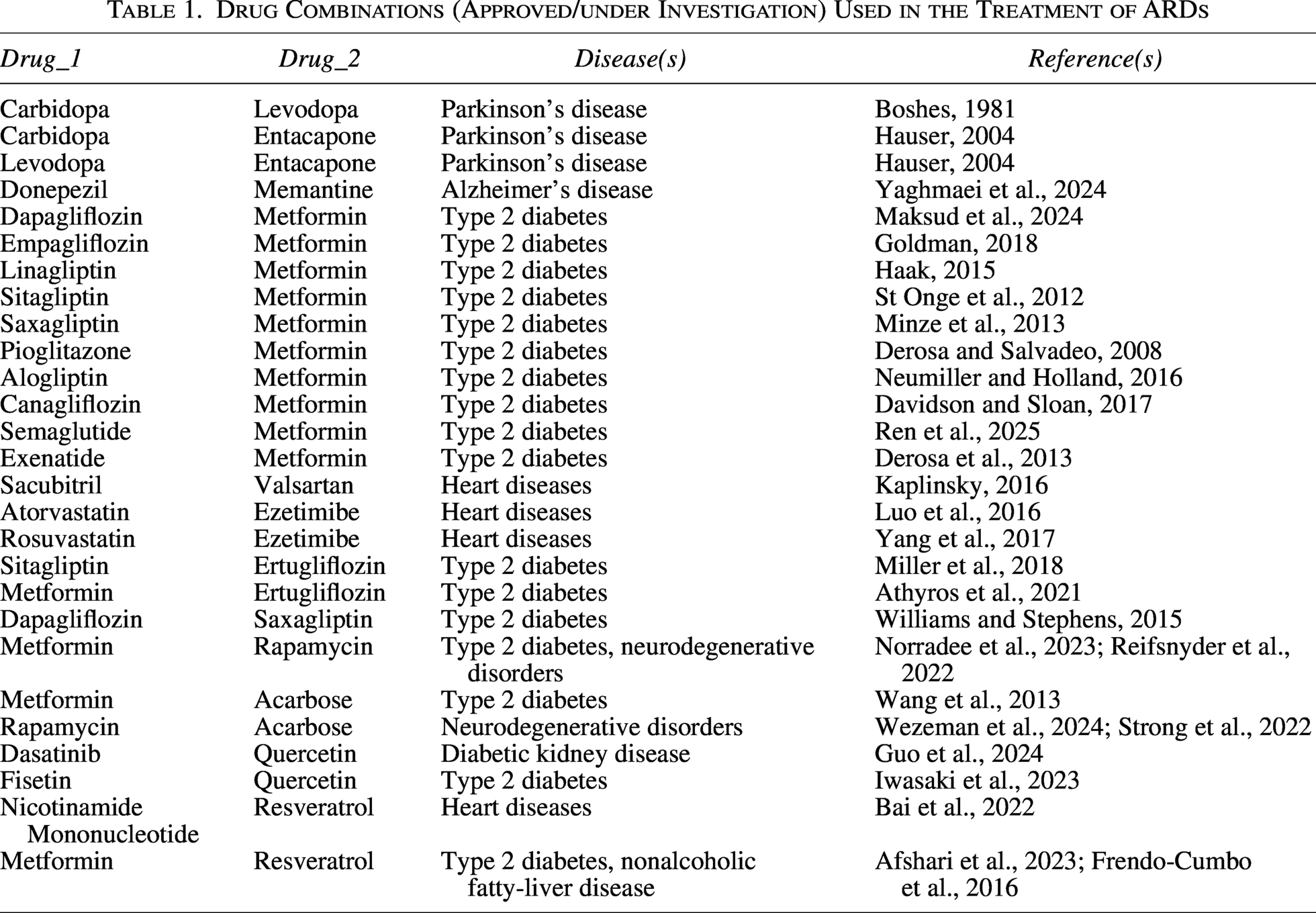
A dataset of 27 drug combinations either approved or under investigation for the treatment of various ARDs was curated (Table 1).
Drug Combinations (Approved/under Investigation) Used in the Treatment of ARDs
Thereafter, 1024-bit molecular fingerprints of each drug in the combination were calculated using RDKit (Landrum and Greg, 2006), which were used as node features and the known combination information as edges to build a GNN-based synergy prediction model in order to predict potential combinations among the screened natural compounds.
GNN-based geroprotective combinations prediction
The graph was constructed using PyTorch Geometric’s Data object (Fey and Lenssen, 2019), with molecular fingerprint vectors as node features and undirected edge indices (from known drug pair combination knowledge). A three-layer graph convolutional network (GCN) was defined to learn node embeddings of the compounds, and an edge prediction model was built on top, incorporating the GCN and a fully connected layer that concatenated embeddings of compound pairs and applied a sigmoid activation to predict the likelihood of an edge between them (edge probability) as synergy probability (Kipf and Welling, 2016). The model was trained using binary cross-entropy loss, optimized with AdamW, and stabilized by early stopping. To guarantee reproducibility, fixed random seeds were applied across NumPy, PyTorch, and CUDA backends (Pedregosa et al., 2011). Rather than selecting negative samples at random, non-synergistic pairs were generated through a hard-negative strategy in which compound pairs were ranked by the cosine similarity of their molecular fingerprints and negatives were drawn from this similarity-based distribution. This ensured that the negative set was chemically informed and avoided trivial random assignment, compelling the model to learn deeper and meaningful relational features. Model’s robustness was evaluated using five-fold stratified cross-validation, reporting accuracy, precision, recall, F1-score, and ROC–area under curve (AUC) values for each fold.
To benchmark the proposed GNN-based synergy prediction framework against a simpler baseline, a similarity-based approach was implemented using cosine similarity between molecular fingerprints of compounds. Specifically, 1024-bit molecular fingerprints were used to compute pair-wise cosine similarity scores between drug pairs, which were then directly treated as proxy scores for synergy likelihood. A threshold-based classification was applied to derive binary predictions and performance was evaluated using standard ROC–AUC. This baseline serves as a chemically intuitive reference model that relies solely on structural similarity, without incorporating network topology or learned representations, thereby enabling assessment of the added value provided by the GNN-based framework.
Furthermore, the model was subjected to external validation using an independently curated dataset of geroprotective drug combinations with experimentally reported therapeutic synergy in various ARD models. This dataset was compiled through an extensive literature review. Molecular fingerprints of the constituent compounds were used as node features, while the experimentally validated combinations were represented as edges. The trained GNN model was then applied to this dataset by embedding each compound into the drug-drug network through connections to structurally similar drugs, enabling integration of new nodes into the existing graph and subsequent generation of node embeddings for the unseen compounds. This approach allows the GNN model to propagate information across the network through message passing and facilitates the generation of context-aware embeddings, which is consistent with prior graph-based frameworks that integrate similarity-driven neighborhood construction with network-based learning (Peng et al., 2022; Zhao et al., 2025). The expanded graph was then processed through the trained GNN model to generate node embeddings, which were subsequently used to compute synergy probabilities for each combination, enabling assessment of its ability to generalize beyond the training data. Once validated, the trained model was applied to the set of screened natural compounds and all possible pairs among them. Each compound was similarly embedded into the drug–drug network by connecting it to structurally similar drugs and the expanded graph was processed through the trained GNN to generate node embeddings. All possible natural compound pairs were then evaluated by the synergy prediction model, producing synergy probability scores for each pair and a highly stringent probability threshold (≥0.999) was applied to identify the most promising natural compound combinations.
Visualization of predicted natural compound interaction network
The graph of natural compound pairs predicted by the GNN-based synergy prediction model was visualized using NetworkX (Hagberg et al., 2008).
Results and Discussion
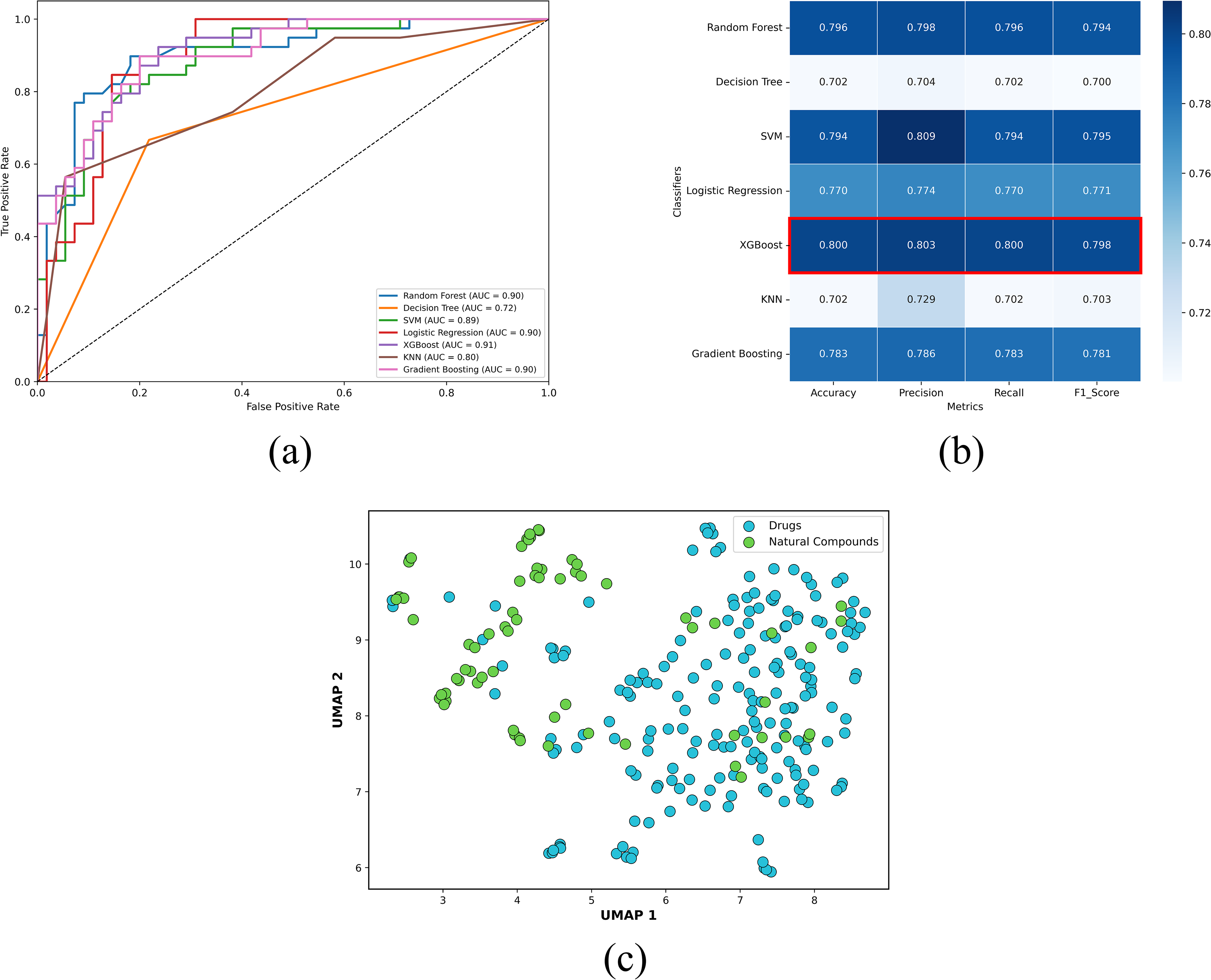
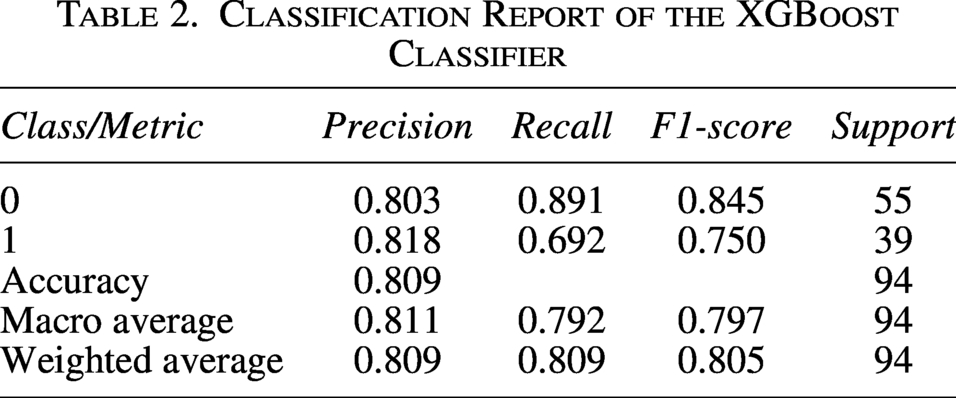
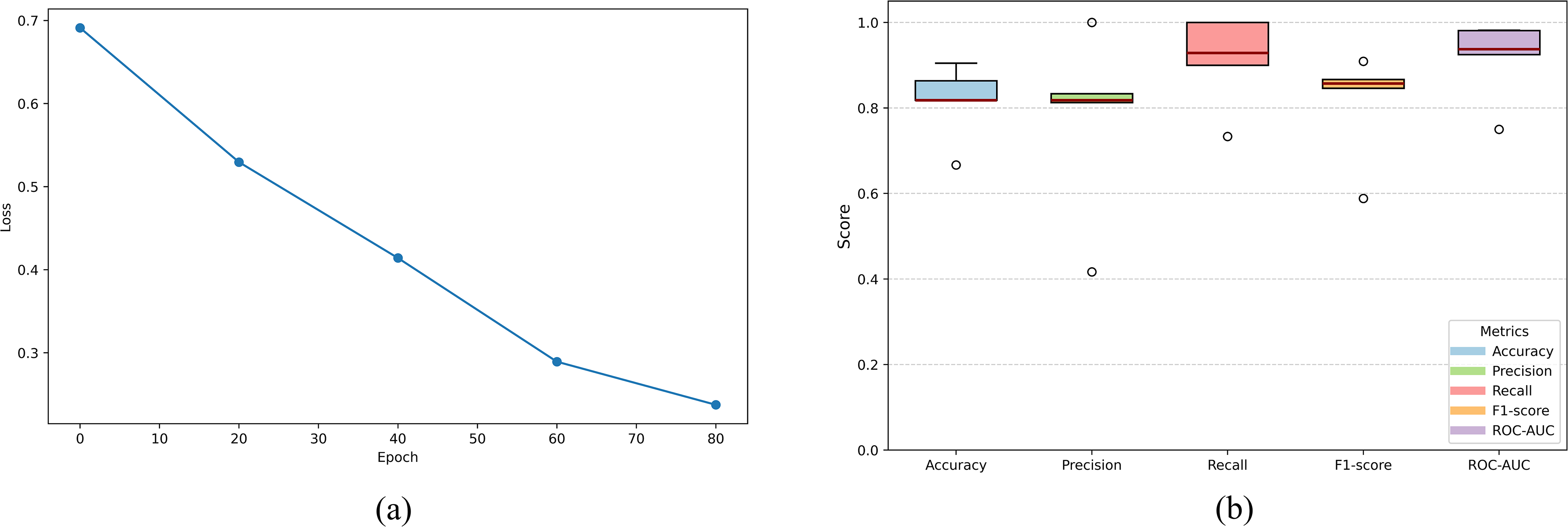
The machine learning-based geroprotector classification model was trained on 1024 molecular fingerprints of FDA-approved drugs for treatment of ARDs, as the positive control set and drugs for NARDs, as the negative control set (Supplementary Table S1). Multiple iterations were conducted for different classifiers, including Random Forest, Decision Tree, SVM, Logistic Regression, XGBoost, KNN, and Gradient Boosting, in order to find an ideal classifier (Chen and Guestrin, 2016; Pedregosa et al., 2011). Consequently, XGBoost emerged as the most effective classifier, achieving an ROC–AUC value of 0.91 and robust performance metrics, indicating strong discriminative ability in distinguishing between the two classes (Fig. 2a and Table 2).
Classification Report of the XGBoost Classifier
In order to validate the model’s (XGBoost classifier) performance, five-fold stratified cross-validation was conducted, which resulted in mean accuracy of 0.800, mean precision of 0.803, mean recall of 0.800, and mean F1-score of 0.798 across folds (Fig. 2b). In addition, the model was evaluated on an independent dataset of experimentally validated geroprotectors retrieved from the Geroprotectors.org database (Moskalev et al., 2015). The model achieved an accuracy of 0.861, correctly identifying 124 out of 144 geroprotectors, with an average confidence score of 0.764 (Supplementary Table S2). Notably, well-established geroprotectors were predicted with significantly high confidence, including Resveratrol (confidence score = 0.977), Quercetin (confidence score = 0.853), and Curcumin (confidence score = 0.976) (Supplementary Table S2) (Proshkina et al., 2024; Rivero-Segura et al., 2024). These results indicate that the model is capable of capturing key features associated with geroprotective activity and maintains predictive consistency on an independent dataset as well, thereby supporting its robustness and applicability for identifying potential geroprotectors. Subsequently, the dataset of 1152 natural compounds, retrieved from PubChem using the “Phytochemicals” search query (Supplementary Table S3), with their molecular fingerprints, was fed to the trained and validated geroprotector classification model. The model predicted 69 natural compounds to be potentially therapeutic for ARDs such as neurological disorders, type 2 diabetes, heart diseases, and chronic kidney disease with a confidence score ≥0.99 (Supplementary Table S4).
To assess potential distribution shift, UMAP was applied to the molecular fingerprints of FDA-approved ARD drugs used for training the classification model and 69 predicted geroprotective natural compounds (Supplementary Tables S1 and S4) (McInnes and Healy, 2018). The projection showed significant overlap between the two groups, with no clearly isolated clusters observed for the natural compounds (Fig. 2c). This indicates that the model operates within a relevant applicability domain and is not extrapolating into entirely unseen chemical regimes, thereby supporting its reliability when applied to structurally distinct natural compounds (Fig. 2c). In addition, several natural compounds were found to be interspersed within the dense clusters of ARD drugs, highlighting the presence of shared feature space and local structural similarity with clinically validated therapeutics (Fig. 2c). Moreover, a subset of natural compounds was observed to occupy adjacent yet continuous regions that remain proximal to the distribution of FDA-approved ARD drugs, which indicates the presence of novel scaffolds of potential therapeutic importance (Fig. 2c). Collectively, these findings demonstrate that the model achieves a balance between generalization and specificity. The observed overlap suggests that many predicted geroprotective natural compounds possess ARD drug-like structural and physicochemical properties, while the adjacent extensions indicate the presence of potential scaffolds of therapeutic significance not widely explored in current ARD therapeutics. These natural compounds were further tested for their drug-likeness using SWISS ADME and 55 out of the 69 natural compounds met the criteria with zero violations of Lipinski’s Rule of Five and were selected for further screening (Daina et al., 2017; Lipinski et al., 2001).
In order to find natural compounds with the ability to simultaneously engage all the targets of interest including Nrf2, mTORC1, SIRT1, and AMPK and potentially, induce a comprehensive and synergistic geroprotective effect, molecular docking was conducted directly with the targets or their regulators. In order to activate Nrf2, molecular docking was conducted with its negative regulator Keap1 at the site involved in its interaction with Nrf2 (Heightman et al., 2019), whereas for mTORC1, docking was done at the site involved in known bisteric inhibitor binding (Burnett et al., 2023) and for AMPK and SIRT1, the molecular docking was done at the site involved in known activator binding (Cameron et al., 2016; Dai et al., 2015). The grid box parameters for each target protein are described in Section Materials and Methods. Subsequently, binding affinities of all 55 natural compounds were obtained (Supplementary Table S5). By applying a binding affinity threshold of ≤ –7.5 for Keap1, mTORC1, and AMPK, 12 natural compounds were identified with comparatively stronger binding affinities across all four targets, suggesting their potential to simultaneously modulate the activity of intricately linked geroprotective regulators (Table 3).
Natural Compounds with Most Significant Binding Affinities with All Four Target Proteins
In addition, site-specific redocking validation was performed to assess the structural plausibility of the predicted interactions. For each target protein, the cocrystallized ligand, including J6Q (inhibitor) for Keap1, XYU (inhibitor) for mTORC1, 6VT (allosteric activator) for AMPK, and 4TO (activator) for SIRT1 was redocked into its respective binding site using the same docking parameters in BIOVIA Discovery Studio (Berman et al., 2000; Burnett et al., 2023; Cameron et al., 2016; Dai et al., 2015; DASSAULT SYSTÈMES, 2025; Heightman et al., 2019). Redocking was performed both individually and in the presence of the 12 screened natural compounds, respectively, for each target protein, in order to confirm the colocalization of natural compounds with the cocrystallized ligand within the same binding pocket of the target protein. The resulting binding poses revealed that these compounds consistently occupied the same functional binding regions as the respective co-crystallized ligands, despite structural diversity (Fig. 3). Notably, the natural compounds exhibited spatial proximity and orientation within the desired sites comparable to the standard/reference ligands, indicating specific and structurally consistent interactions (Fig. 3). These findings support the potential of the identified natural compounds to significantly engage with the target proteins and provide a rational basis for further experimental validation to confirm these computationally derived inferences.
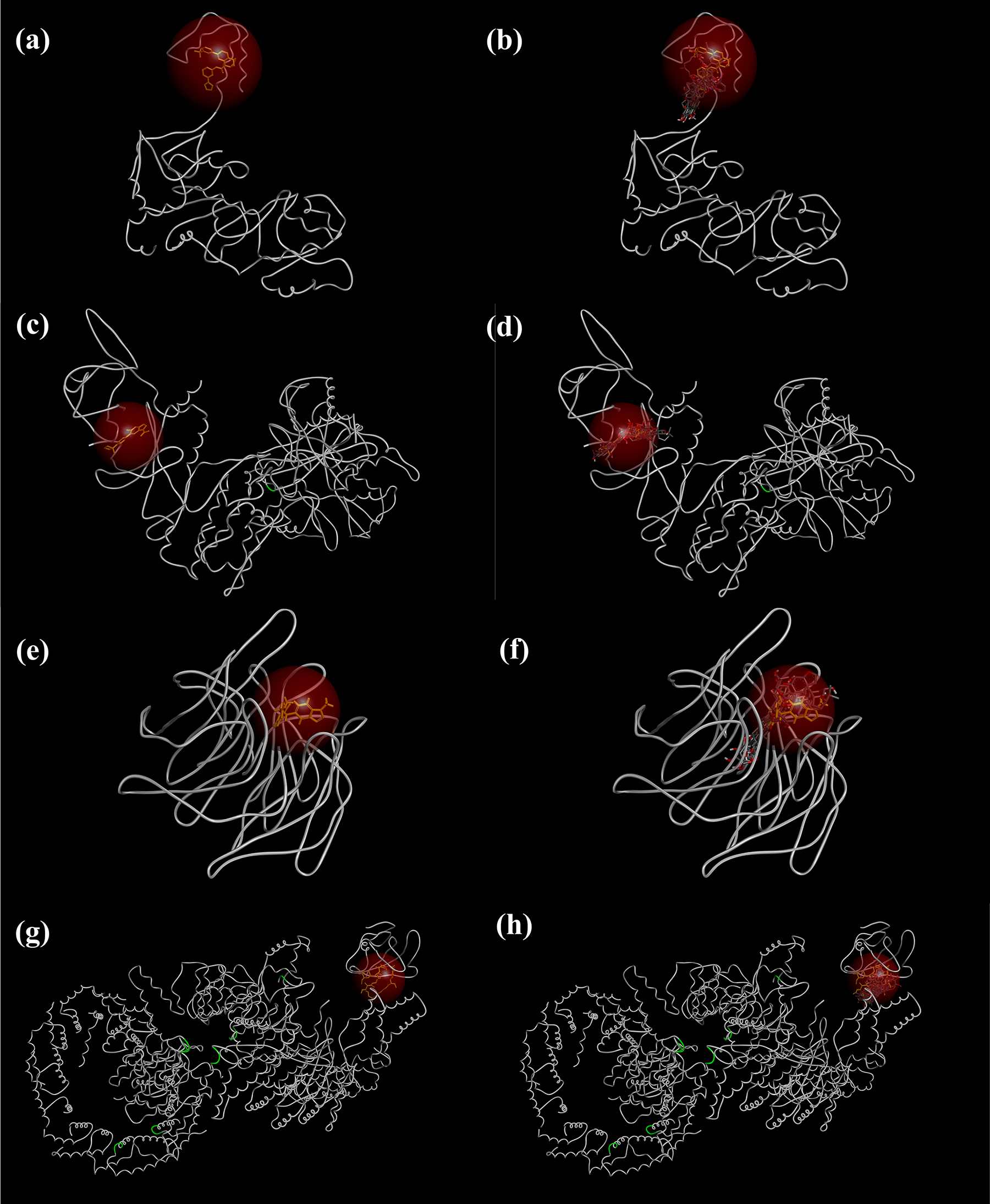
Docking validation showing co-localization of the 12 screened natural compounds with co-crystallized ligand within the binding pocket of the target protein.
Identified as potential geroprotectors by a well-trained and validated classification model, these 12 natural compounds exhibited zero violations of Lipinski’s Rule of Five, indicating favorable drug-like properties. Subsequent molecular docking analysis revealed their potential to regulate the core factors implicated in various ARDs. Taken together, these computational findings position the identified natural compounds as candidates with the potential to simultaneously engage key geroprotective factors and modulate their interconnected network, to ultimately induce a comprehensive and coordinated therapeutic effect across multiple ARD models, subject to further experimental validation.
Owing to the importance of combinatorial strategies, particularly in therapeutic interventions focused on multiple cross-regulatory target engagement, such as the one presented here, the study further explored potential geroprotective combinations among the screened 12 natural compounds using GNN (GCN, in particular) (Kipf and Welling, 2016). A GNN model was trained on 27 drug combinations either approved or under investigation for treatment of various ARDs (Table 1), where 1024-bit molecular fingerprints calculated for each individual drug were provided as node features and known combination information was used as edges, consequently generating 27 such edges. As discussed earlier, the GNN model was trained to predict synergy probabilities and was evaluated periodically for loss convergence using binary cross-entropy loss. The model showed a consistent decrease in loss over the epochs and subsequently the loss flattened close to zero, suggesting convergence and thus indicating effective learning and model stability (Fig. 4a). This consistent minimization of loss suggests that the model successfully captured the underlying structural patterns of compound-compound interactions. The model was further evaluated for its robustness and reliability via five-fold stratified cross-validation, where the model displayed consistent performance across each fold and achieved noteworthy performance metrics, including mean accuracy of 0.8143 ± 0.0806, precision of 0.7761 ± 0.1927, recall of 0.9124 ± 0.0978, F1 score of 0.8135 ± 0.1146 and ROC-AUC of 0.9151 ± 0.0856 (Fig. 4b). In order to further validate the model’s robustness, an independent dataset was curated through a comprehensive literature review, consisting of 15 experimentally validated combinations reported to induce significant therapeutic effects in various ARD models. The GNN-based synergy prediction model was then applied to this independent dataset and it correctly identified 11 out of 15 geroprotective combinations with a synergy probability ≥ 0.872 for each predicted combination (Table 4). This level of recovery indicates that the model is capable of capturing key ARD-specific interaction patterns and generalizing beyond the training dataset, thus supporting its applicability in computationally identifying potential synergistic combinations for various ARDs.
External Validation of the GNN-Based Synergy Prediction Model Using an Independent Dataset of Experimentally Validated Geroprotective Combinations across Multiple ARD Models.
The table lists compound pairs along with their predicted synergy probabilities and supporting literature references. The model successfully identified 11 out of 15 combinations with high synergy probabilities (≥0.872), demonstrating its ability to capture core ARD-specific interaction patterns and generalize beyond the training dataset
Despite its relatively simple architecture and limited training data, the model was able to identify several experimentally validated combinations with high confidence and showed consistent performance across cross-validation and external validation datasets. This suggests that the learned node embeddings were able to capture meaningful structural and relational patterns within the drug-drug interaction network. By leveraging a graph convolutional architecture, the model encodes relationships between compounds through both feature representation and network structure. While more advanced synergy prediction models utilize larger datasets (Liu et al., 2024; Torkamannia et al., 2023), the current results indicate that even a relatively lightweight GNN-based approach can capture relevant interaction patterns between compounds when guided by appropriate structural representations and network context. To further contextualize model performance, a similarity-based baseline was evaluated using cosine similarity between molecular fingerprints as a proxy for synergy prediction. The baseline approach yielded substantially lower performance (ROC-AUC = 0.555), indicating limited discriminative ability when relying solely on chemical similarity (Supplementary Fig. S1). In contrast, the GNN-based framework achieved a markedly higher ROC-AUC (0.915), suggesting that the proposed approach is able to capture interaction patterns that are not readily reflected by structural similarity alone (Supplementary Fig. S1). Collectively, these findings highlight the potential of GNN-based frameworks as tools for exploring synergistic interactions even in data-constrained settings, supporting their practical utility in early-stage computational screening, particularly in scenarios where large-scale datasets are not readily available. Nevertheless, as a computational framework, this approach requires further validation through experimental studies to further enhance its reliability in early-stage screening, particularly in comparison to more data-intensive models.
Following successful training and validation, the GNN-based synergy prediction model was applied to the dataset of 12 natural compounds and all 66 possible combinations of compound pairs were generated for synergy probability prediction. The model successfully predicted the synergy probabilities between all the natural compound pairs (Supplementary Table S6). To identify high-confidence predictions, a stringent screening threshold of synergy probability ≥ 0.999 was applied, which yielded five natural compound pairs involving four distinct natural compounds, including Baicalein, Pectolinarigenin, Phloretin and Demethoxycurcumin (Table 5).
Top Natural Compound Pairs with Synergy Probability ≥0.999

The graph of all 66 natural compound pairs formed using synergy probabilities predicted by the GNN-based synergy prediction model was visualized using NetworkX, where synergy probabilities were used as edge weights (Fig. 5).
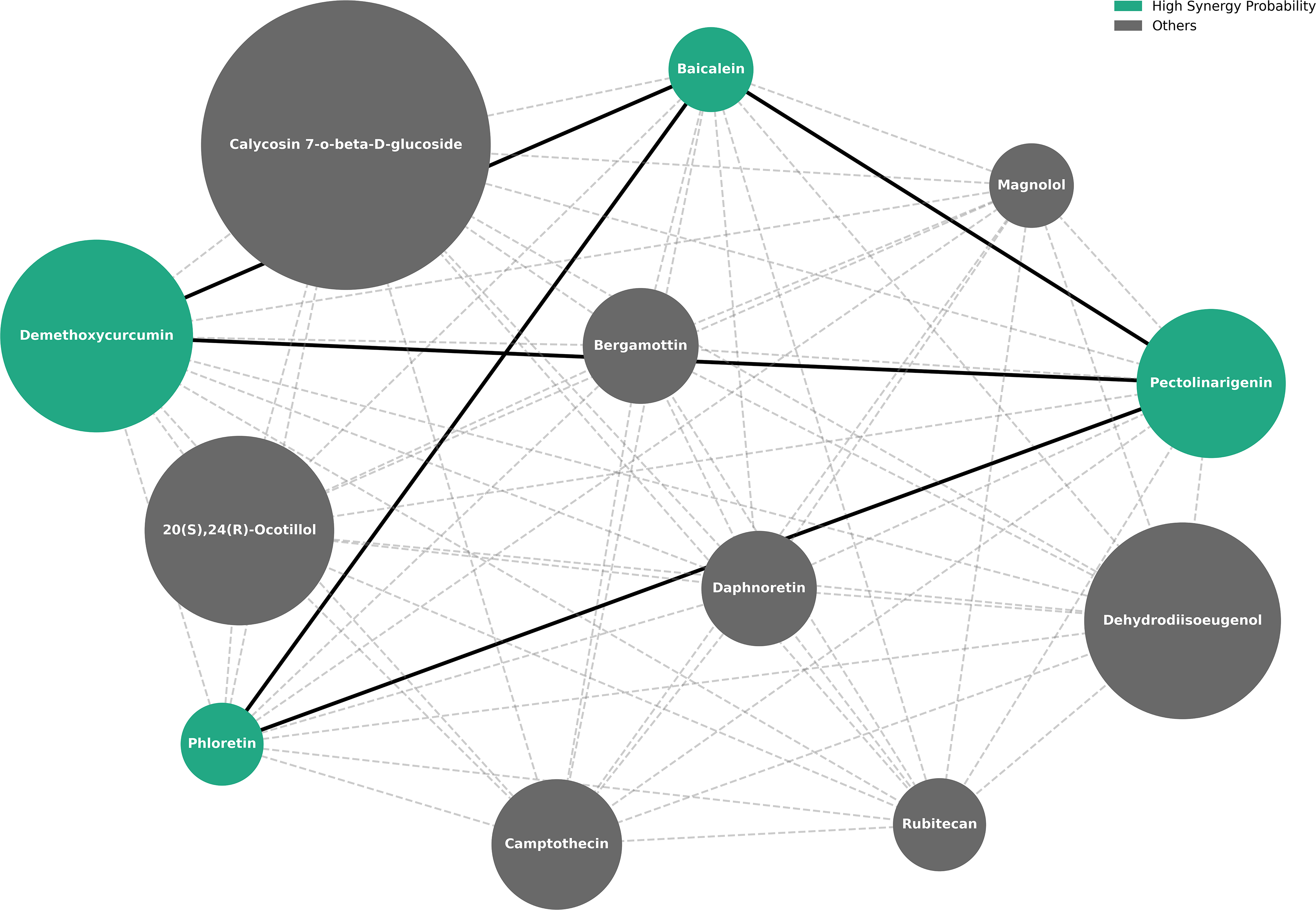
Graph visualization of predicted natural compound combinations. Nodes shown in green represent compound pairs with a synergy probability ≥0.999, connected by solid edges, while the remaining nodes are shown in gray and are connected by dashed edges, indicating lower predicted synergy.
Conclusion
This study presents a systems biology-informed computational framework for the identification of natural compound combinations with the potential to modulate multiple interconnected geroprotective regulators across diverse ARDs. By integrating machine learning-based compound classification, molecular docking for multi-target interaction assessment and GNN-based synergy prediction, the proposed framework provides a systematic and scalable approach for early computational screening of geroprotective candidates and their potential combinations. The present study identified 12 natural compounds as promising candidates with computationally predicted geroprotective capability, favorable drug-like properties and the ability to interact with multiple targets of therapeutic significance, including Nrf2, mTORC1, AMPK, and SIRT1. While these findings suggest possible multi-target engagement, the proposed effects on cellular processes such as oxidative stress regulation, autophagy, proteostasis, and mitochondrial function remain computationally inferred and require experimental validation. To further investigate combinatorial potential, a GNN-based synergy prediction model was employed to identify potential geroprotective combinations among the screened natural compounds. Despite a relatively small dataset and simple architecture, the model demonstrated consistent performance across cross-validation and external validation and was able to recover several high-confidence combinations during validation, suggesting that the learned embeddings capture ARD-specific interaction patterns within the network, likely driven by the GCN-based encoding of relationships between compounds through both feature representation and network structure.
Nevertheless, a set of high-confidence natural compound combinations was identified using this framework, involving Baicalein, Pectolinarigenin, Phloretin, and Demethoxycurcumin. These combinations align with the proposed multi-target geroprotective framework and show the potential to elicit comprehensive and coordinated geroprotective effects. Future improvements could be achieved by incorporating larger datasets and in vitro/in vivo validation to test biological efficacy for enhanced robustness and reliability of our approach.
Authors’ Contributions
Y.S.: Conceptualization, data curation, methodology, writing—original draft. A.D.: Supervision, writing—review and editing, validation.
Data Availability Statement
All data supporting the findings of this study are included within the article and its Supplementary Data. Additional related data are available from the corresponding author upon reasonable request.
Supplemental Material
sj-docx-1-omi-10.1177_15578100261464019 — Supplemental material for Synergistic Geroprotectors Mapping through Systems Machine Learning and Graph Neural Networks
Supplemental material, sj-docx-1-omi-10.1177_15578100261464019 for Synergistic Geroprotectors Mapping through Systems Machine Learning and Graph Neural Networks by Yuvraj Sharma, and Asmita Das
Supplemental Material
sj-xlsx-2-omi-10.1177_15578100261464019 — Supplemental material for Synergistic Geroprotectors Mapping through Systems Machine Learning and Graph Neural Networks
Supplemental material, sj-xlsx-2-omi-10.1177_15578100261464019 for Synergistic Geroprotectors Mapping through Systems Machine Learning and Graph Neural Networks by Yuvraj Sharma, and Asmita Das
Supplemental Material
sj-xlsx-3-omi-10.1177_15578100261464019 — Supplemental material for Synergistic Geroprotectors Mapping through Systems Machine Learning and Graph Neural Networks
Supplemental material, sj-xlsx-3-omi-10.1177_15578100261464019 for Synergistic Geroprotectors Mapping through Systems Machine Learning and Graph Neural Networks by Yuvraj Sharma, and Asmita Das
Supplemental Material
sj-xlsx-4-omi-10.1177_15578100261464019 — Supplemental material for Synergistic Geroprotectors Mapping through Systems Machine Learning and Graph Neural Networks
Supplemental material, sj-xlsx-4-omi-10.1177_15578100261464019 for Synergistic Geroprotectors Mapping through Systems Machine Learning and Graph Neural Networks by Yuvraj Sharma, and Asmita Das
Supplemental Material
sj-xlsx-5-omi-10.1177_15578100261464019 — Supplemental material for Synergistic Geroprotectors Mapping through Systems Machine Learning and Graph Neural Networks
Supplemental material, sj-xlsx-5-omi-10.1177_15578100261464019 for Synergistic Geroprotectors Mapping through Systems Machine Learning and Graph Neural Networks by Yuvraj Sharma, and Asmita Das
Supplemental Material
sj-xlsx-6-omi-10.1177_15578100261464019 — Supplemental material for Synergistic Geroprotectors Mapping through Systems Machine Learning and Graph Neural Networks
Supplemental material, sj-xlsx-6-omi-10.1177_15578100261464019 for Synergistic Geroprotectors Mapping through Systems Machine Learning and Graph Neural Networks by Yuvraj Sharma, and Asmita Das
Supplemental Material
sj-xlsx-7-omi-10.1177_15578100261464019 — Supplemental material for Synergistic Geroprotectors Mapping through Systems Machine Learning and Graph Neural Networks
Supplemental material, sj-xlsx-7-omi-10.1177_15578100261464019 for Synergistic Geroprotectors Mapping through Systems Machine Learning and Graph Neural Networks by Yuvraj Sharma, and Asmita Das
Footnotes
Acknowledgments
The present study has been carried out in the Department of Biotechnology, Delhi Technological University, following all ethical principles of the university. The authors acknowledge the Junior Research Fellowship (JRF) awarded to Y.S. by Council of Scientific and Industrial Research (CSIR), India.
Author Disclosure Statement
Authors declare no conflict of interest.
Funding Information
The authors declare that no funding or grants were received to support the conduct of this study.
Abbreviations Used
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.