
Abstract
The spread of a virus can be modeled as the diffusion of virion populations along the edges of a host contact network. Most prior research assumes that the virus maintains the same genome throughout the diffusion process, and, consequently, the genome itself is of no modeling interest. Eletreby et al. do consider a model of multiple variants in which a virus can mutate within a host into another variant before transmission across an edge. Still, they fail to model the genome or any established fitness models. We incorporate three biological notions, fitness landscapes, viral quasispecies, and genome structure, to more accurately model the diffusion of a virus with mutations. We investigate established fitness landscape models and various simulated contact networks. We simulate the diffusion process across the contact network, incorporating co-occurring evolutionary and epidemiological processes. In our proof-of-concept simulations, while determining which variant should infect an exposed individual, we consider the fitness values of the variants. ViraFit tunes the ruggedness of the fitness landscape by varying several attributes: genome length, network size, mutation rate, infection probability, and infection time. Our simulation results demonstrate the importance of including the fitness value of each variant and how the fitter ones persist over time.
INTRODUCTION
The spread of a viral infection in a host population is commonly modeled as a diffusion process on a contact network, where nodes represent hosts and edges represent opportunities for transmission (Leventhal et al., 2015; Eletreby et al., 2020). In most such models, the virus is treated as a fixed entity: once introduced, the pathogen propagates across the network without meaningful genetic change, or mutation is assumed to be negligible relative to the timescale of transmission. This assumption is often violated in practice. Many RNA viruses, including SARS-CoV-2, undergo rapid mutation within hosts, generating genetically diverse populations during the course of an outbreak (Domingo, 1998, 2006). Consequently, viral diffusion and viral evolution are not independent processes but unfold concurrently.
Recent work has begun to relax the single-strain assumption. Notably, Eletreby et al. (2020) introduced a multi-strain epidemic model in which a virus may mutate within a host before being transmitted to others. Their framework allows competition among strains during transmission and captures the co-occurrence of epidemiological and evolutionary dynamics. However, this and related models (Alexander and Day, 2010; Leventhal et al., 2015) represent strains abstractly and do not explicitly model genome structure, mutational neighborhoods, or established notions of evolutionary fitness. As a result, they cannot capture how specific mutations, genome length, or the organization of sequence space shape the emergence and persistence of viral variants.
In contrast, biological evolution is fundamentally governed by the relationship between genotype and fitness. A fitness landscape associates each genomic variant with a measure of reproductive success, thereby defining which mutations are beneficial, neutral, or deleterious (Fragata et al., 2019). When mutations occur one site at a time, variants form the nodes of a high-dimensional sequence space (a hypercube), and evolution proceeds as an adaptive walk along edges connecting neighboring genotypes (Kauffman and Weinberger, 1989). Classical landscape models, such as House-of-Cards (HoC) (Kauffman and Levin, 1987), NK (Kauffman and Weinberger, 1989), and Rough Mount Fuji (Neidhart et al., 2014), describe different degrees of ruggedness and epistasis, shaping the number of local optima and the accessibility of fitter variants. These models provide a principled framework for studying evolutionary dynamics; yet, they have rarely been integrated into network-based epidemic models.
Moreover, viral evolution is best understood not at the level of a single genotype, but as a population of closely related variants. RNA viruses typically exist as quasispecies: dynamic mutant swarms generated by high replication error rates, in which individual genomes differ by one or a few nucleotides from one another (Domingo, 2006; Domingo and Perales, 2019). Despite continual mutation, these populations often display remarkable phenotypic stability, reflecting strong selection operating over structured sequence space rather than random drift (Leitmeyer and Rico-Hesse, 1997). Representing viral populations as isolated strains, therefore, obscures the evolutionary mechanisms that govern adaptation, persistence, and competition.
Finally, viral genomes possess diverse lengths and architectures that directly affect mutational neighborhoods and evolutionary potential. RNA viruses range from compact genomes of a few kilobases to complex genomes exceeding
Taken together, these observations reveal a fundamental gap in existing network epidemic models: while they capture the spread of infection, they largely ignore genome-level evolution, the structure of sequence space, and fitness-driven selection. Although prior work allows mutation among abstract strains (Eletreby et al., 2020), it does not incorporate explicit genotype representations, fitness landscapes, or quasispecies dynamics. As a result, such models cannot explain how specific mutations arise, how fitter variants outcompete others, or how evolutionary trajectories depend jointly on genome properties and network structure.
In this work, we introduce
The primary novelty of ViraFit lies in modeling viral evolution at the level of explicit genomes rather than as a fixed set of abstract strains. Existing network-based epidemic models either assume a single, immutable pathogen or represent multiple variants as discrete types with predefined mutation or transmissibility matrices (Alexander and Day, 2010; Eletreby et al., 2020). Such approaches do not capture the structure of sequence space, the mutational neighborhood of a genome, or the emergence of fitness from genotype. Conversely, classical fitness landscape models describe adaptive walks in genotype space but ignore population-level transmission on contact networks. ViraFit unifies these two perspectives by embedding viral genomes as nodes in a hypercube sequence space, associating each genotype with a fitness value, and coupling within-host mutation to between-host transmission on a network. This enables the study of how genome length, mutational accessibility, landscape ruggedness, and contact topology jointly shape strain emergence, competition, and persistence—phenomena that cannot be represented by constant-strain or matrix-based multi-strain models.
Using established fitness landscape models and a range of synthetic contact networks, we demonstrate that both evolutionary outcomes and epidemic dynamics depend critically on genome length, mutation rate, infection probability, infection duration, and network structure. Our results show that explicitly incorporating fitness alters diffusion patterns and that fitter variants consistently emerge and persist over time. By bridging genotype-level evolution with network-based transmission, ViraFit provides a biologically grounded framework for studying viral adaptation during outbreaks. ViraFit is publicly available at https://github.com/Badhan023/Viral_Fitness_Landscape.
BACKGROUND
Classical fitness landscape models provide standard mechanisms for controlling landscape ruggedness and epistasis, which, in turn, shape evolutionary accessibility and adaptive walk behavior. We summarize three widely used models: HoC, NK, and Rough Mount Fuji, to (1) situate ViraFit within established landscape theory and (2) motivate the design of our explicit, tunable genome–fitness function. In our implementation, these models can additionally be used as optional baseline landscape generators for comparison against the proposed fitness formulation (Section 3.1).
Several models have been introduced to visualize the concept of the fitness landscape. Wright (1932) presented the fitness landscape as a high-dimensional map, typically in three dimensions, in which genotypes are plotted in the
In the NK model (Kauffman and Weinberger, 1989), fitness landscapes are defined by three parameters: N, A, and K. For a protein or genome sequence, N denotes the number of loci (sequence length), A denotes the number of possible states per locus (alphabet size), and K specifies the number of other loci whose states epistatically influence the contribution of a given locus to overall fitness. Thus, K controls the degree of epistasis in the system and is conceptually equivalent to the ruggedness of the fitness landscape: when
The Rough Mount Fuji model (Neidhart et al., 2014) introduces a genotype-fitness model that combines a random HoC landscape with an additive landscape. In the simplest version of the model, the additive selective advantage, s, is the same for all loci. By varying s relative to the standard deviation of the HoC fitness values, the ruggedness of the landscape can be tuned (de Visser and Krug, 2014).
METHODS
ViraFit has two main parts: genome-fitness and network models, which portray the viral evolution fitness landscape. The genome-fitness model represents the quasispecies environment comprising all strains of a particular viral genome, whereas the network model represents the contact network among hosts. The overall model is obtained by combining these two models: the simulation of mutation and infection operates on the network model, while the genotype-fitness model influences the network.
Genome-Fitness model
Let us consider that we have a viral genome S of length N. There exist four nucleotides, A (Adenine), T (Thymine), C (Cytosine), G (Guanine), and T (Thymine) [U (Uracil) in RNA]. As a result, the initial genome will have one of
Since it is difficult to picture such high-dimensional spaces, let us consider a genome of length N constrained to use two nucleotides, A and G, to compose the structure of the hypercube. Let 1 and 0 represent A and G, respectively; each strain is a binary string of length N. Then, such binary strings can be easily defined as vertices of an N-dimensional Boolean hypercube (Kauffman and Levin, 1987), where the number of vertices will be
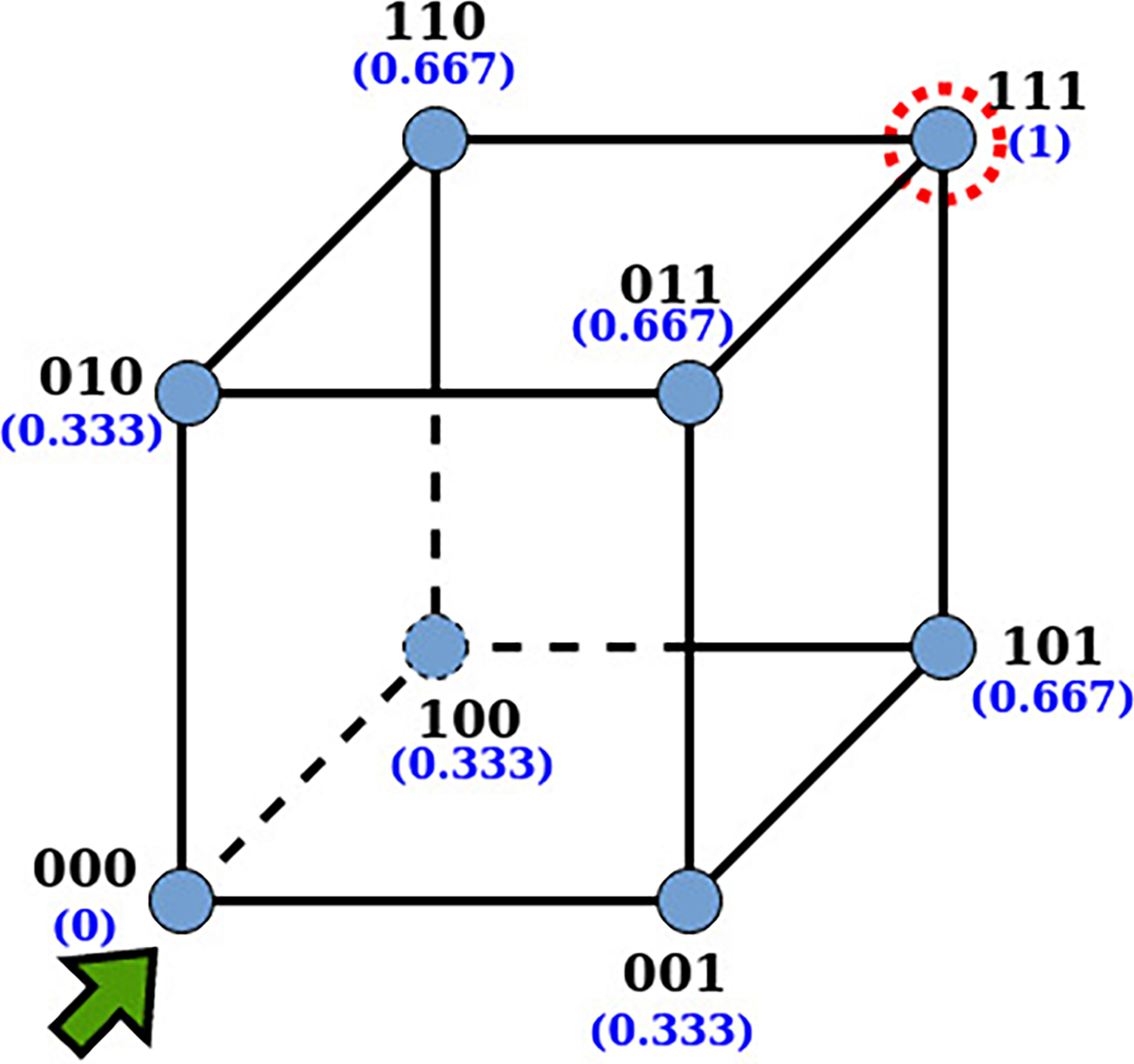
A three-dimensional Boolean hypercube, in which each vertex represents one of the possible binary sequences. Each strain has three neighbors and a fitness value in parentheses. The fittest strain is 111, marked by a dotted red circle; the least fit strain is 000, from where our simulation starts.
Each vertex of the hypercube H has a fitness value, F, associated with it. The fitness function is designed as Eqn. 1 to keep it simple. Each hypercube node has N neighbors, since each nucleotide has two options: 0 or 1.
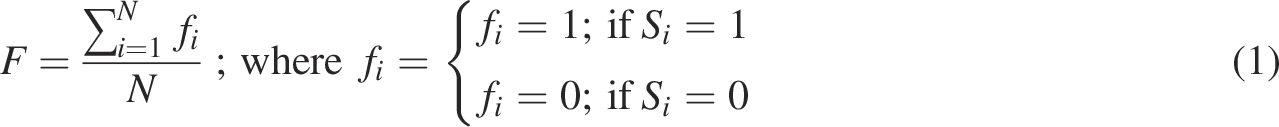
For computational tractability, we represent each genomic position using a reduced alphabet that preserves the structure of the mutational neighborhood while enabling large-scale simulation. This simplification does not affect the qualitative behavior of the adaptive process, which is governed by the fitness landscape topology.
Let

In the Waxman model, each node has only one attribute: the position in the coordinate plane. We have modified the Waxman model by adding two other features to each node to prepare the graph for the epidemiology network: infection status and infection duration. The status of a node can be uninfected(inactive) if a node has not received any infection or infected (active) of strain type i, for
Unless otherwise stated, parameters not explicitly varied in the experiments (e.g., the simulated annealing schedule T,
Each simulation proceeds in discrete iterations. In each iteration, ViraFit performs: (1) within-host mutation in genotype space (a walk on the hypercube H governed by fitness and simulated annealing), (2) infection decision for exposed susceptible hosts based on both neighborhood prevalence and strain fitness, and (3) infection/recovery updates based on the infection duration parameter
Adaptive-walk statistics
We define adaptive walk length as the number of simulation iterations required for the globally fittest strain in the predefined fitness landscape (hypercube H) to first appear in the host population. Because the fitness landscape is fixed and identical across all simulation replicates, this definition ensures direct comparability of adaptive walk length across different epidemiological and network parameter settings.
Algorithm
At the beginning of the simulation, we started with the least fit strain,
The full algorithm,
Parameters Used for the Model and Their Description

Parameters Used for the Model and Their Description
To determine the fitness of mutant strains produced during within-host evolution, we employ a simulated annealing procedure that governs how mutations move across the fitness landscape. The simulated annealing algorithm (Bertsimas and Tsitsiklis, 1993) is a probabilistic technique for approximating the global optimum of a given function, inspired by physical annealing. It is an effective and general method for finding global optima in the presence of many local optima. The idea is that this algorithm starts from the annealing temperature, T, and loops through the iterations needed to cool down to the minimum temperature
In every iteration, every infected host has a probability of mutating; see Figure 2. The mutation rate

Pseudocode for updating infected nodes.

Pseudocode for simulated annealing to select the next strain, if any.
Pseudocode for returning the fitness value of a strain.
See Figures 5 and 6. In
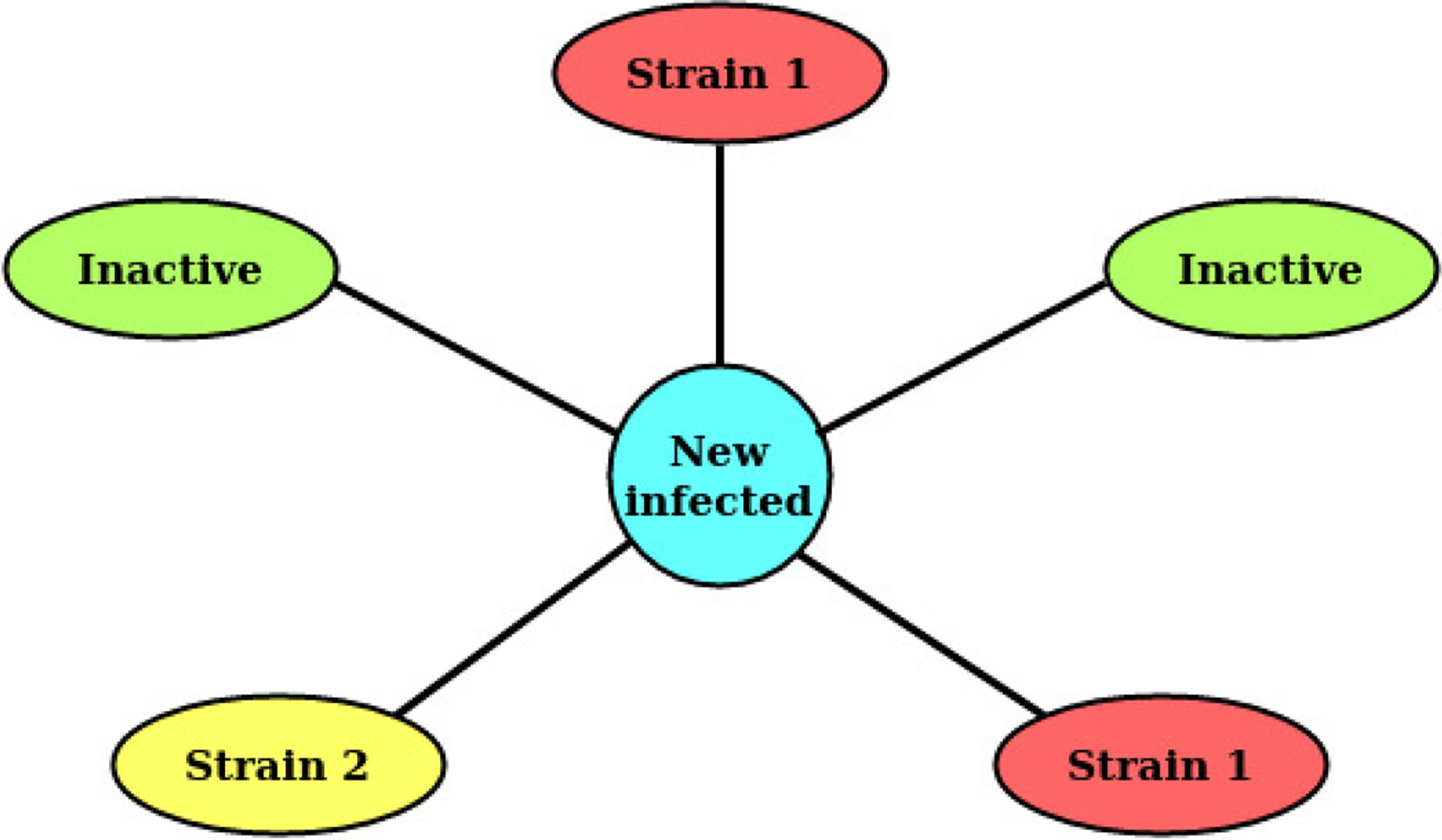
Decision making at the moment of a new infection. The host that is going to be infected is the one in the middle, and there are five neighbors, among which two are inactive, two are infected by strain 1, and the other one by strain 2.

Contact-driven infection decision. Infection probability increases with the number of infected neighbors,

Pseudocode for infection and recovery to update the timer and status of the infected nodes, and change the timer and status of the new infected nodes.

Pseudocode for infection propagation.
Algorithm 6 describes the procedure. Let v be an uninfected host, and let 
If infection occurs, the transmitted strain is selected probabilistically from among the strains present in 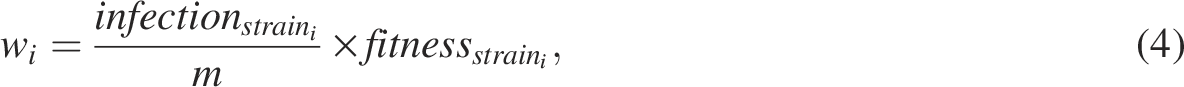

Importantly, while
Finally, the infection decision is formulated at the host level. The quantity
This is the third phase in which the timer and the status of each node in G are updated. Each host node has its own timer set to 0 while uninfected. Once a node is infected, the timer starts and increments by 1 in each iteration. When it reaches the maximum duration limit, this algorithm resets it to 0 and the status to “uninfected”. A node is considered recovered when its timer reaches the duration
Infection propagation
The
RESULTS
In this section, we examine how key evolutionary and epidemiological parameters in ViraFit influence the adaptive walk toward the globally fittest genotype, starting from the least-fit initial strain. The adaptive walk length is defined as the number of iterations required for the globally fittest genotype in the fixed hypercube fitness landscape to first appear in the host population.
ViraFit integrates parameters from both the fitness landscape and the host contact network. Evolutionary parameters include genome length and mutation rate (
Unless otherwise specified, default values are
Effects of mutation rate, genome length, infection probability, and infection duration
Figure 9a illustrates the effect of mutation rate on the time required to reach the globally fittest genotype. Across all network sizes, lower mutation rates are associated with longer adaptive walk lengths, indicating slower traversal of the fitness landscape. Increasing
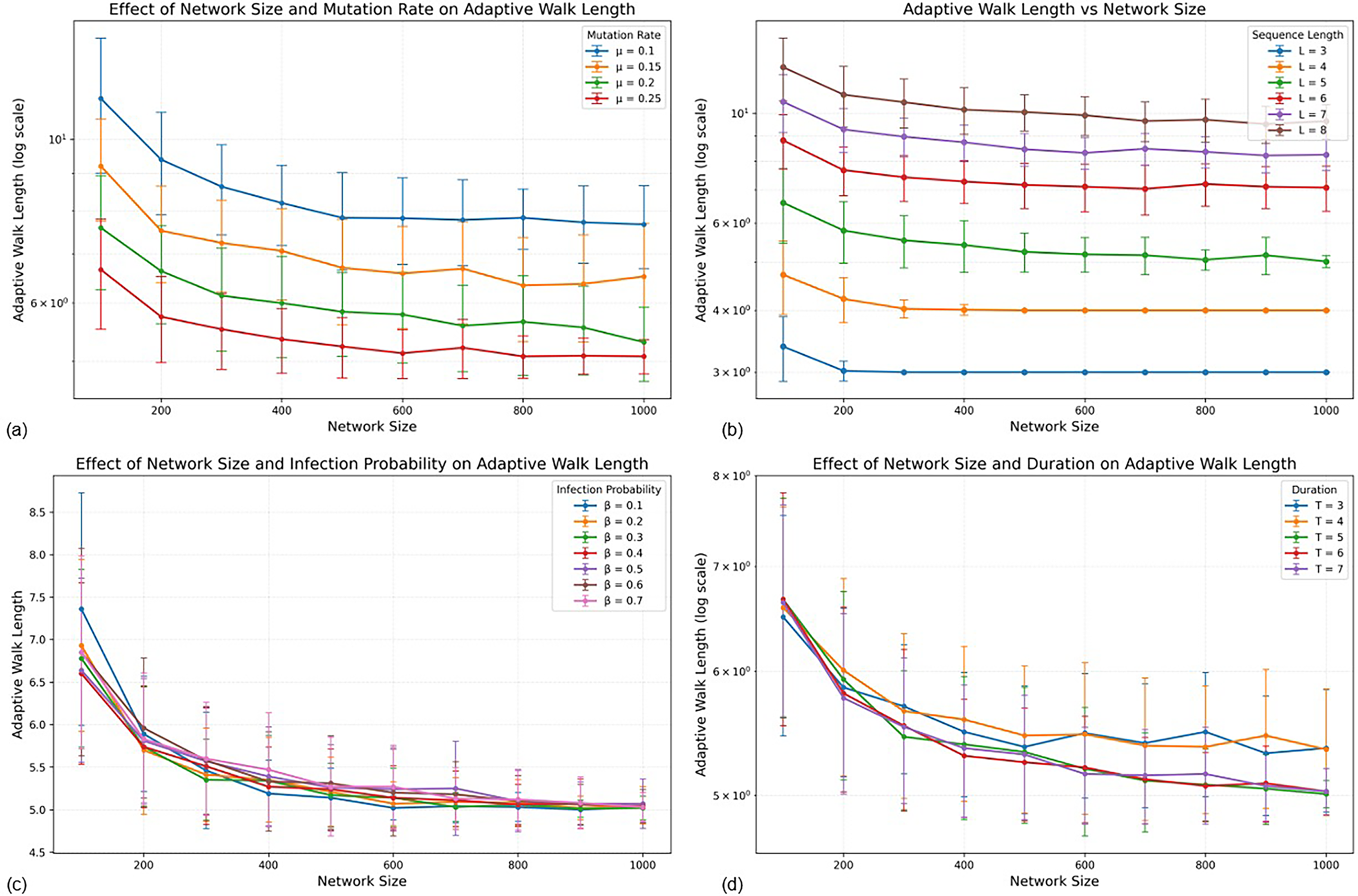
The interaction between genome length and network size is shown in Figure 9b. Shorter genomes exhibit consistently shorter adaptive walks across all network sizes, reflecting the smaller size and lower dimensionality of the corresponding fitness landscape. As genome length increases, walk length increases monotonically, consistent with increased landscape dimensionality and mutational search space. Adaptive walk length varies smoothly with network size, without abrupt transition points. Larger networks generally lead to modest increases in walk length for longer genomes, suggesting that increased host population size enables broader exploration of genotype space before fixation of the global optimum.
The effect of infection probability
Figure 9d shows the effect of infection duration. Shorter infection durations are associated with slightly longer adaptive walks, particularly in larger networks. Longer durations provide an extended within-host evolutionary opportunity before recovery, thereby increasing the probability that fitter variants arise and propagate. However, the magnitude of this effect is smaller than that observed for the mutation rate.
Overall, the mutation rate remains the dominant determinant of convergence speed to the global fitness peak. Genome length controls the dimensional complexity of the fitness landscape and therefore the overall magnitude of adaptive walk length. Epidemiological parameters primarily modulate the rate at which advantageous variants diffuse through the contact network rather than fundamentally altering evolutionary trajectories.
We systematically evaluated the effects of key parameters governing both the fitness landscape and the contact network, including genome length, mutation rate (
Additional parameters, such as the simulated annealing temperature schedule (T,
Count of infections per strain
We observe that the counts of infections per strain differ. Moreover, not all the strains can persist over time (iterations). For genotype length = 5 and network size = 500, there can be 32 possible strains, as shown in Figure 10. The figure shows the trend in infection counts over iterations, with strains sorted by increasing fitness value.
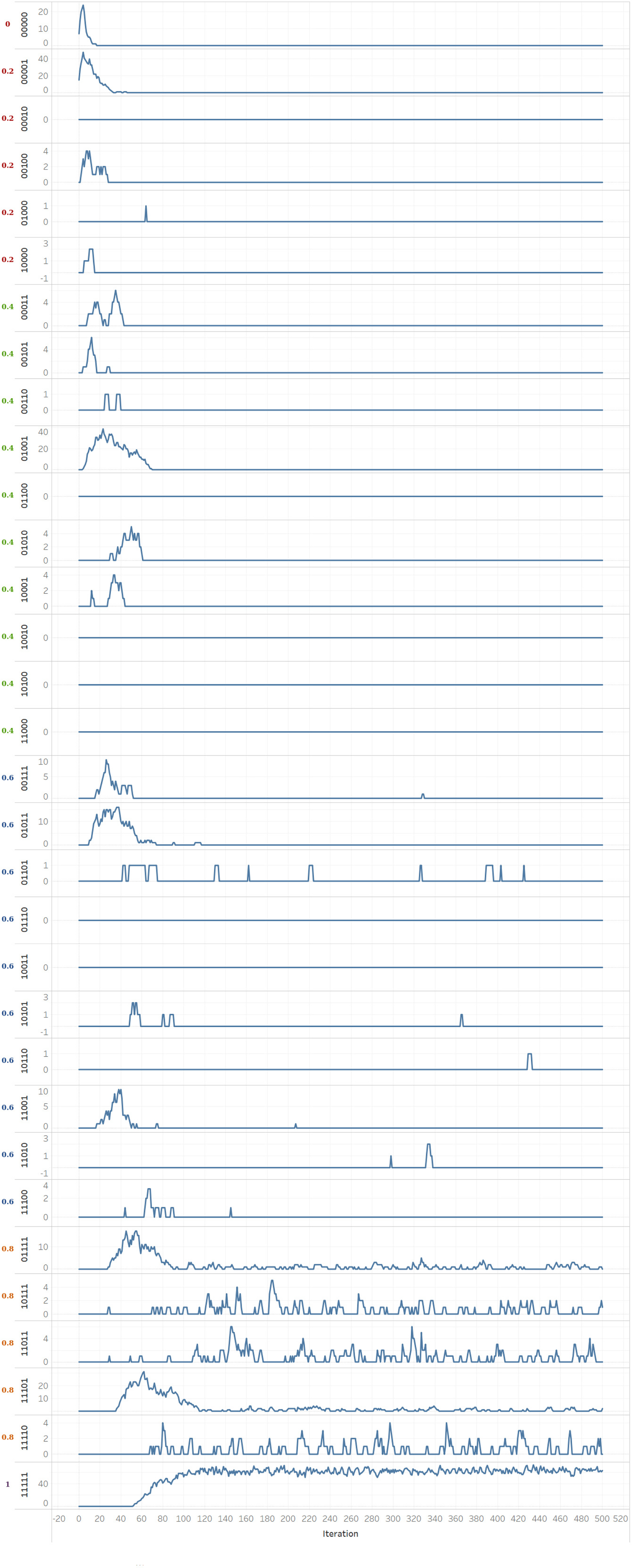
Trends in the count of infections per strain for genotype length = 5 and network size = 500. Here, the strains are organized according to their fitness value, mentioned on the left of each strain. The fitter strains, i.e., 01111, 10111, 11011, 11101, 11110, and 11111, are quite stable and persist over time.
We have also run our model on nucleotide sequences. We simulated only
Table for Genome Length Versus Network Size, Where the Value Shows the Average Fitness Reached for 100 Walks
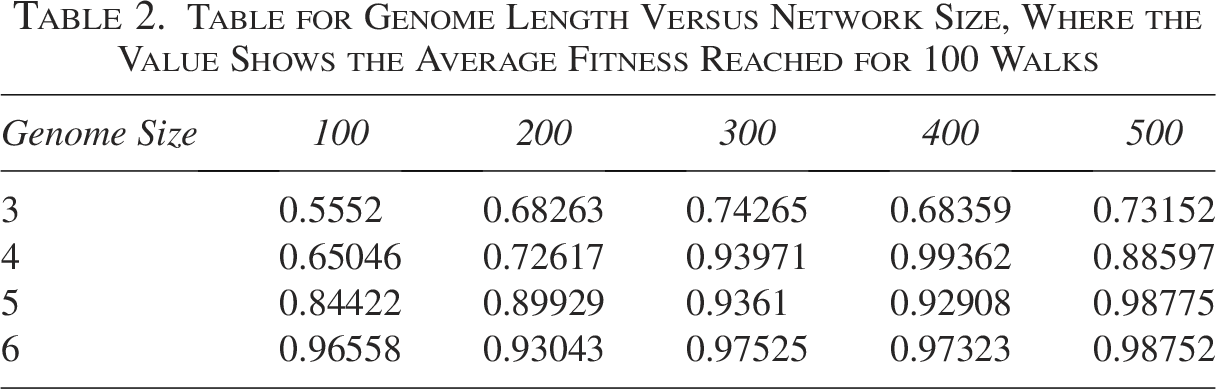
Table for Genome Length Versus Network Size, Where the Value Shows the Average Fitness Reached for 100 Walks
ViraFit provides a unified framework for exploring how viral evolution and epidemiological spread interact through both genome-level fitness landscapes and host contact networks. The model exposes several tunable parameters in both the fitness and network components, allowing systematic investigation of how evolutionary trajectories depend on genetic architecture and population structure. In the fitness model, the form of the fitness function directly shapes the adaptive walk of viral strains. While prior landscape models typically assign fitness values using randomized functions, we adopt a simple formulation to maintain computational tractability. Nonetheless, our framework readily accommodates empirically derived fitness values (de Visser and Krug, 2014), thereby enabling more biologically realistic representations of ruggedness when modeling large-scale nucleotide or protein sequences.
During mutation, a modified simulated annealing procedure (Bertsimas and Tsitsiklis, 1993) governs movement through the fitness landscape. In high-dimensional sequence spaces, where each strain has many mutational neighbors, approximate optimization is essential. Simulated annealing allows the model to favor fitness-increasing mutations while retaining a nonzero probability of accepting fitness-decreasing steps, thereby reflecting the stochasticity inherent in real evolutionary processes. The parameters governing this procedure, the mutation rate
Importantly, our results demonstrate that network structure itself modulates evolutionary dynamics. Although it is expected that the fitness landscape influences which variants spread through the network, we find that network properties, such as size, also affect how strains traverse the landscape. For example, adaptive walks are not identical across networks of different sizes, nor are they invariant when network size is held constant, and sequence length is varied. This indicates that the fitness and network models independently and jointly shape both evolution and infection propagation. As shown in Eq. (4), infection outcomes depend on both local strain prevalence and fitness. Consequently, a less fit strain can dominate transmission if it is sufficiently abundant in the vicinity of a host, even in the presence of fitter competitors. Larger networks amplify this effect by increasing opportunities for such competitive interactions, effectively increasing landscape ruggedness and enabling reversals in adaptive trajectories. Thus, reaching a high-fitness strain does not guarantee its persistence: a population may transiently access a fitness peak yet fail to maintain it under continued competitive pressure.
In ViraFit, strain fitness represents competitive (intrahost) fitness rather than direct per-contact transmissibility between hosts. The probability that an exposed host becomes infected is governed by the epidemiological parameter
When infection occurs, strain selection is probabilistic and based on prevalence-weighted fitness. Specifically, strains present among infected neighbors are assigned weights proportional to their local prevalence and fitness, and the transmitted strain is sampled accordingly. This formulation allows both evolutionary advantage and local abundance to shape transmission outcomes without assuming deterministic competitive exclusion. Consequently, in the absence of competition, strains with different fitness values spread at similar rates determined by
This probabilistic formulation better reflects the stochastic nature of real transmission processes, where infection events arise from independent exposure attempts and less-fit strains may occasionally establish infection due to chance or local abundance effects. At the same time, the model preserves a clear separation between epidemiological transmissibility (governed by
Compared with prior epidemic network models that assume a fixed pathogen or represent multiple strains as abstract types without explicit genomic structures or fitness landscapes (Alexander and Day, 2010; Eletreby et al., 2020), ViraFit introduces a fundamentally different paradigm. Each variant is represented as a point in a structured sequence space (hypercube), and fitness is assigned at the genotype level. Mutation is therefore modeled explicitly as movement through neighboring genomes, rather than as transitions among a predefined set of strain types. This distinction enables ViraFit to capture how genome length, mutational neighborhoods, and landscape ruggedness mechanistically influence evolutionary outcomes—capabilities absent from existing multi-strain network models based solely on mutation or transmissibility matrices.
In this study, adaptive walk length is defined relative to the globally fittest genotype in the fixed fitness landscape. This ensures that walk lengths are directly comparable across parameter settings and reflect the time required to reach the global evolutionary optimum under different epidemiological and network conditions. It quantifies the extent of adaptive exploration permitted by a given epidemiological and network configuration. Longer adaptive walks indicate that the pathogen population can access more distant, potentially higher-fitness peaks in the underlying landscape, rather than implying slower evolutionary dynamics.
CONCLUSION
ViraFit provides a mechanistic framework that integrates genotype-level fitness landscapes with explicit host contact networks to study the joint dynamics of viral evolution and epidemiological spread. By representing strains as vertices in a structured sequence space and coupling mutation, fitness-guided selection, and contact-driven transmission, the model captures how population structure and evolutionary pressure interact to shape adaptive trajectories. The infection formulation ensures that transmission risk increases with local exposure, while fitness-weighted strain selection governs competitive establishment, enabling consistent comparison of adaptive walk lengths across parameter settings.
In practice, multiple strains may share identical or near-identical fitness values, giving rise to numerous local optima in the fitness landscape. Consequently, adaptive walks initiated from different starting genotypes may converge to distinct local or global peaks. Incorporating empirically derived fitness landscapes (de Visser and Krug, 2014) would further enhance biological realism and allow application to specific viral systems.
At present, the model abstracts several epidemiological processes to maintain tractability. Interactions among co-circulating viruses, host immunity, isolation behavior, and disease-induced mortality are not explicitly represented, despite their potential influence on transmission and evolutionary trajectories. Extending ViraFit to incorporate these mechanisms represents an important direction for future work and would enable a more comprehensive investigation of coupled evolutionary–epidemiological systems.
Footnotes
AUTHORS’ CONTRIBUTIONS
Lsh conceived of the project, while BD wrote the software, performed the experiments, and reported the results. Both authors wrote the manuscript and read it thoroughly.
ACKNOWLEDGMENT
This material is based upon work supported by the National Science Foundation under Grant No. CCF-1918656.
AUTHOR DISCLOSURE STATEMENT
No competing financial interests exist.
FUNDING INFORMATION
Funding was provided by the National Science Foundation under Grant No. CCF-1918656.